

Par-4/THAP1 complex and Notch3 competitively regulated pre-mRNA splicing of CCAR1 and affected inversely the survival of T-cell acute lymphoblastic leukemia cells
- PMID: 23975424
- PMCID: PMC3898485
- DOI: 10.1038/onc.2013.349
Par-4/THAP1 complex and Notch3 competitively regulated pre-mRNA splicing of CCAR1 and affected inversely the survival of T-cell acute lymphoblastic leukemia cells
Abstract
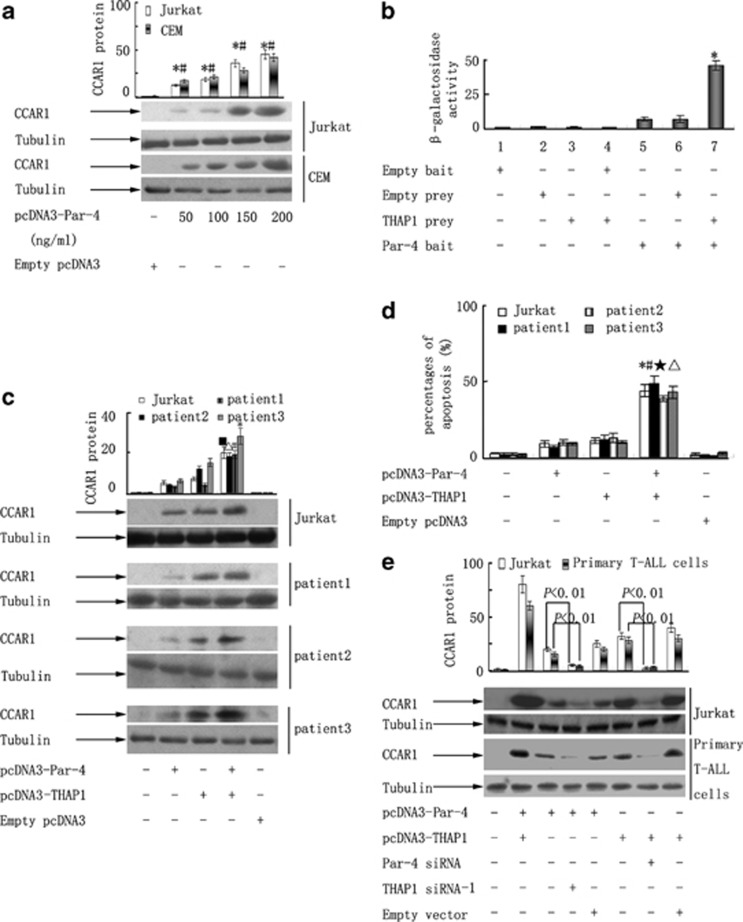
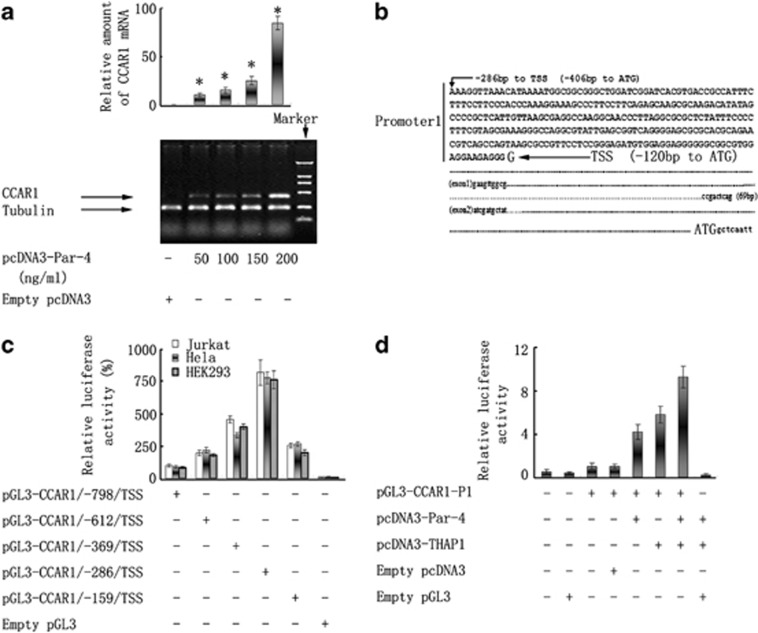
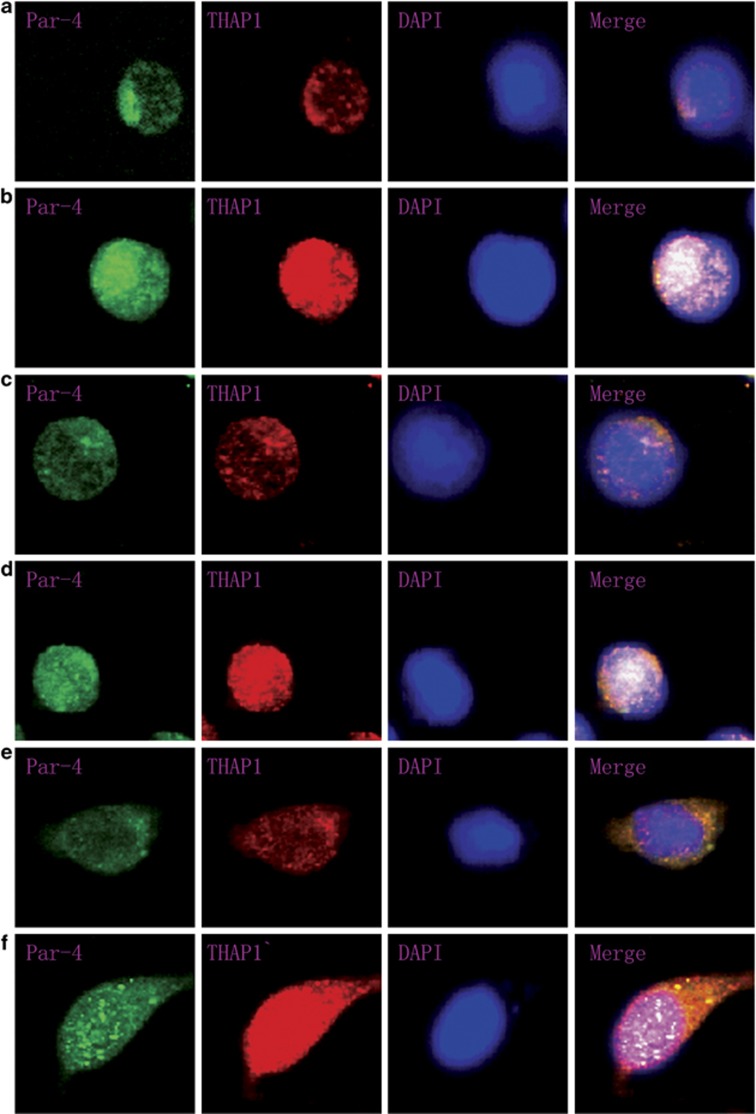
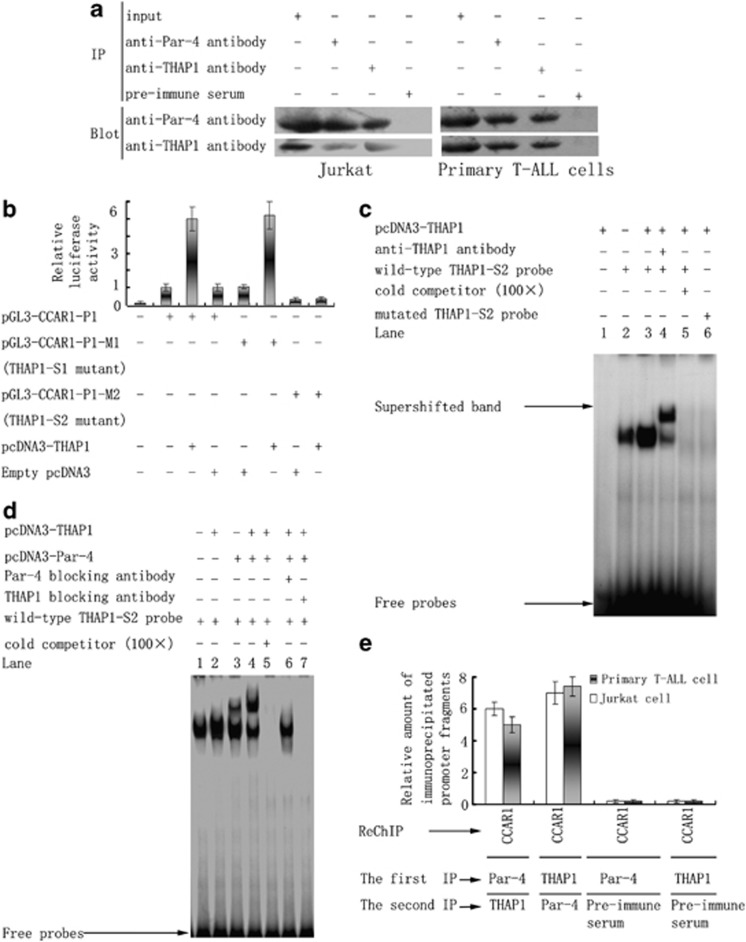
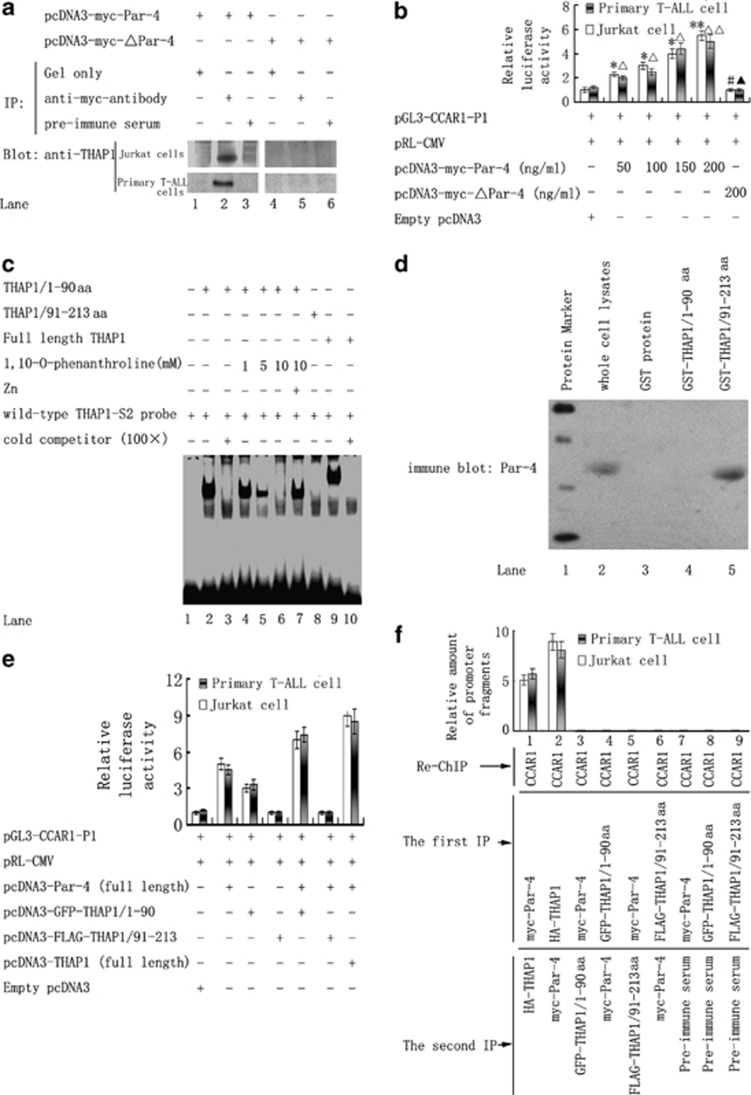
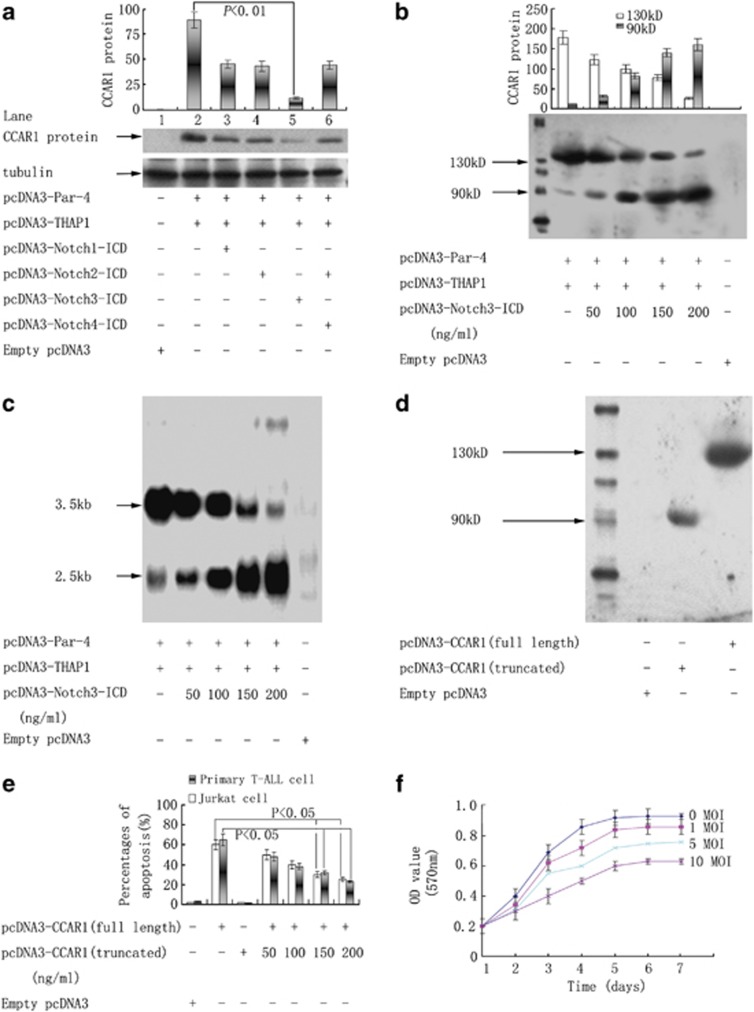
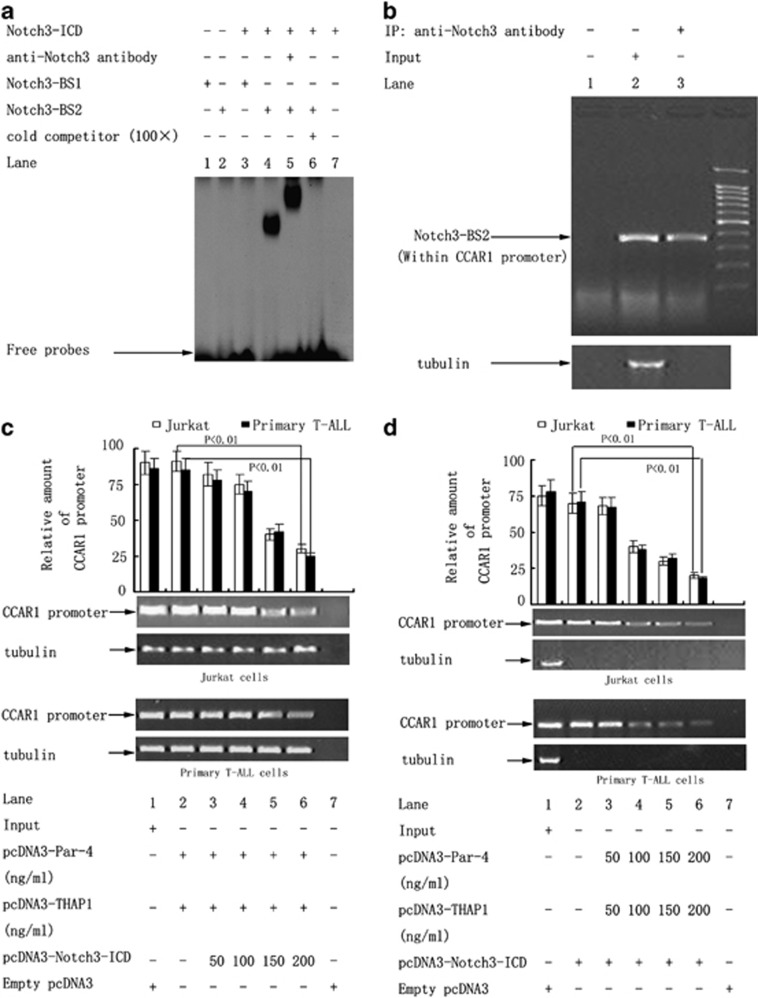
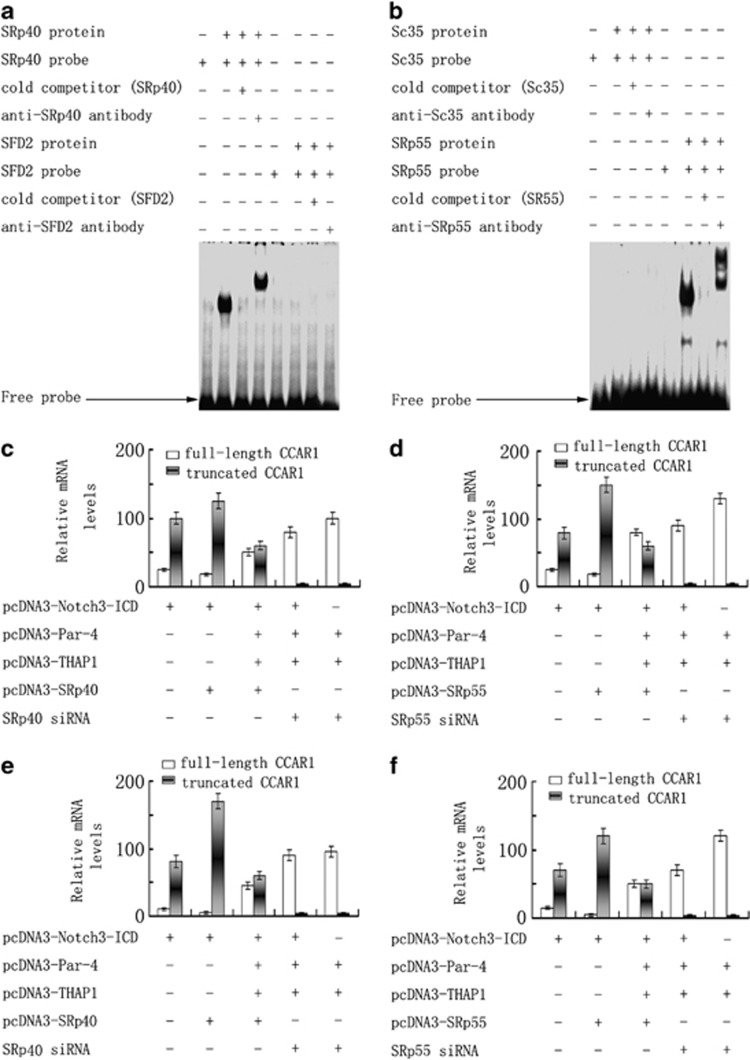
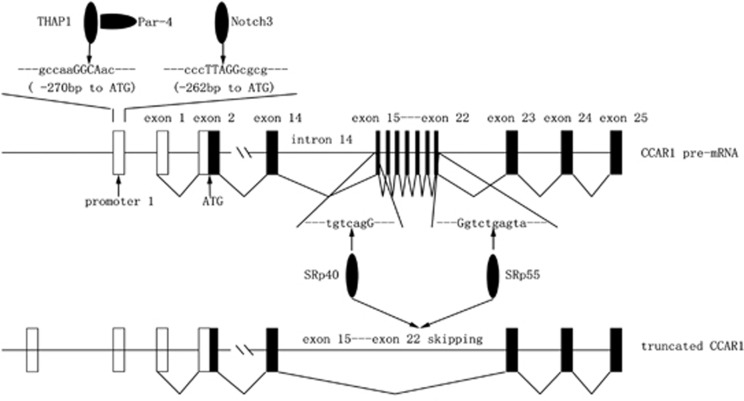
Although the intensification of therapy for children with T-cell acute lymphoblastic leukemia (T-ALL) has substantially improved clinical outcomes, T-ALL remains an important challenge in pediatric oncology. Here, we report that the cooperative synergy between prostate apoptosis response factor-4 (Par-4) and THAP1 induces cell cycle and apoptosis regulator 1 (CCAR1) gene expression and cellular apoptosis in human T-ALL cell line Jurkat cells, CEM cells and primary cultured neoplastic T lymphocytes from children with T-ALL. Par-4 and THAP1 collaborated to activate the promoter of CCAR1 gene. Mechanistic investigations revealed that Par-4 and THAP1 formed a protein complex by the interaction of their carboxyl termini, and THAP1 bound to CCAR1 promoter though its zinc-dependent DNA-binding domain at amino terminus. Par-4/THAP1 complex and Notch3 competitively bound to CCAR1 promoter and competitively modulated alternative pre-mRNA splicing of CCAR1, which resulted in two different transcripts and played an opposite role in T-ALL cell survival. Despite Notch3 induced a shift splicing from the full-length isoform toward a shorter form of CCAR1 mRNA by splicing factor SRp40 and SRp55, Par-4/THAP1 complex strongly antagonized this inductive effect. Our finding revealed a mechanistic rationale for Par-4/THAP1-induced apoptosis in T-ALL cells that would be of benefit to develop a new therapy strategy for T-ALL.
Figures
References
-
- Glienke W, Chow KU, Bauer N, Bergmann L. Down-regulation of wt1 expression in leukemia cell lines as part of apoptotic effect in arsenic treatment using two compounds. Leuk Lymphoma. 2006;47:1629–1638. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
