

Modulation of lipid metabolic defects rescues cleft palate in Tgfbr2 mutant mice
- PMID: 23975680
- PMCID: PMC3857953
- DOI: 10.1093/hmg/ddt410
Modulation of lipid metabolic defects rescues cleft palate in Tgfbr2 mutant mice
Abstract
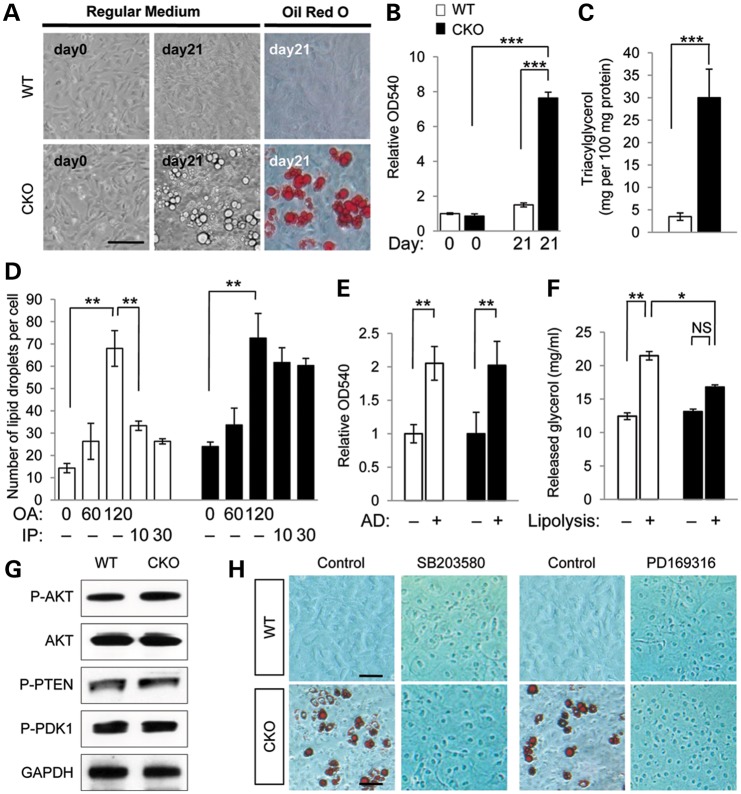
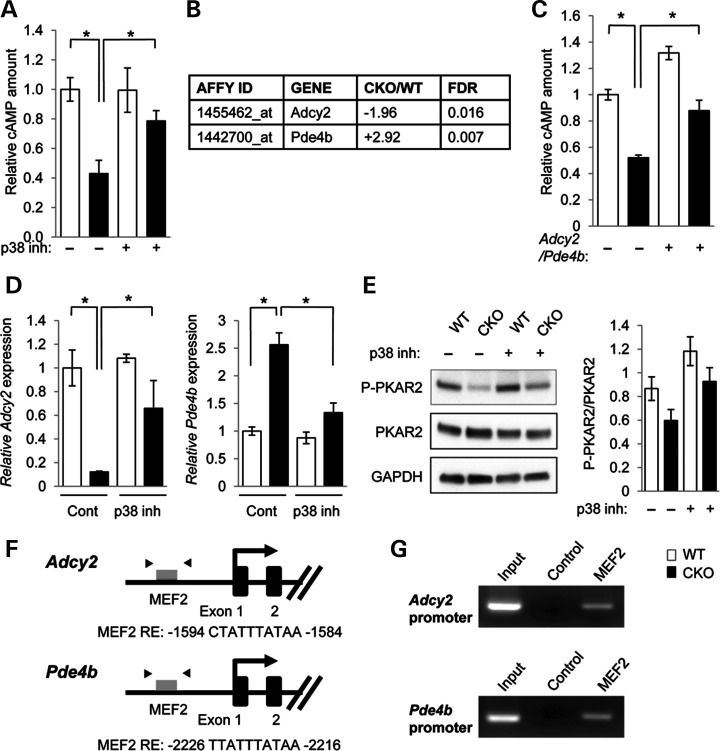
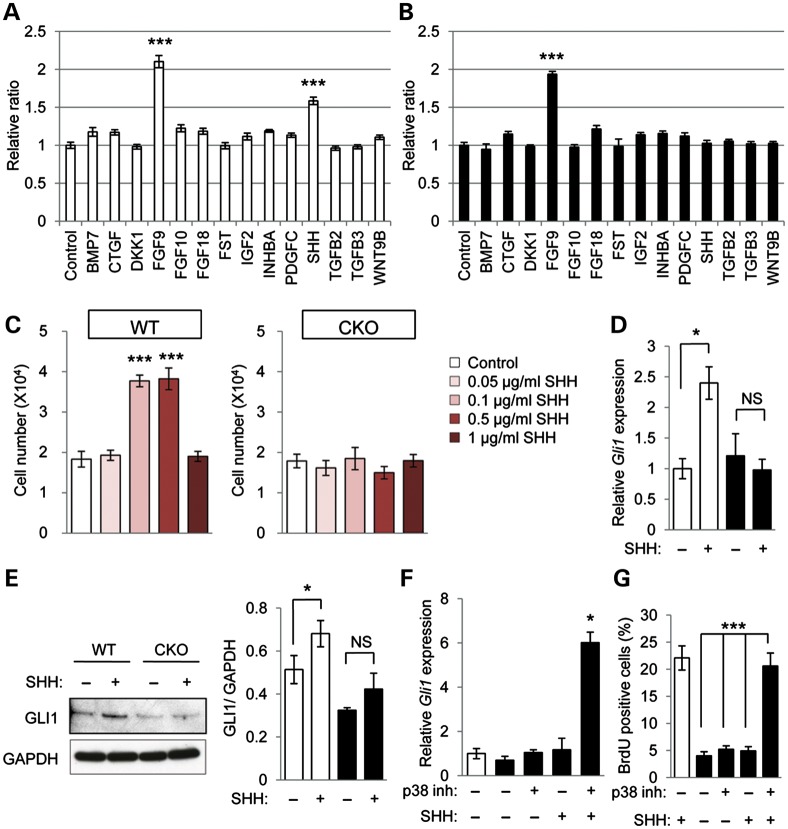
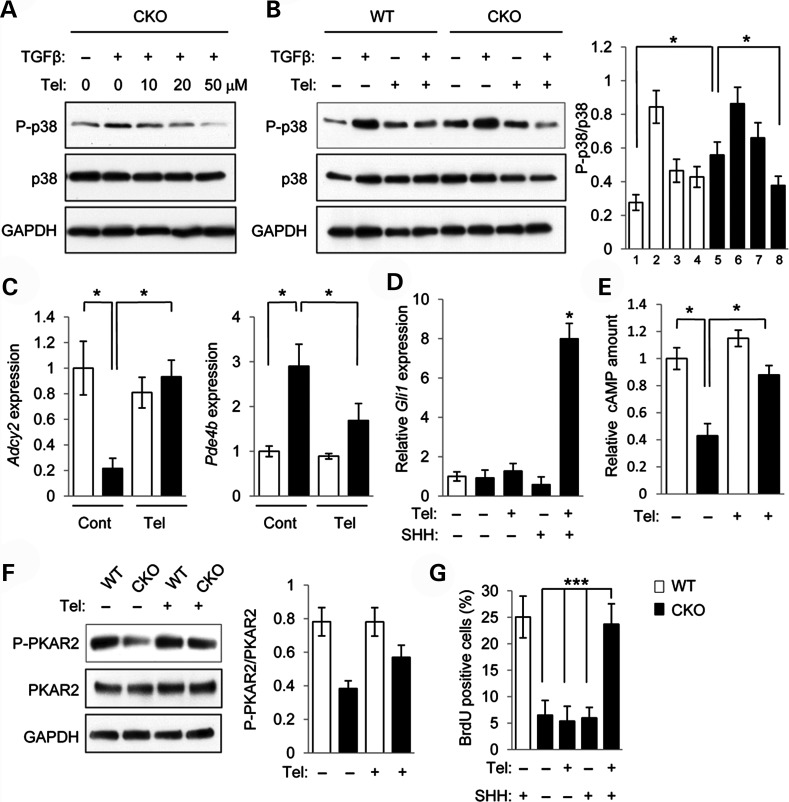
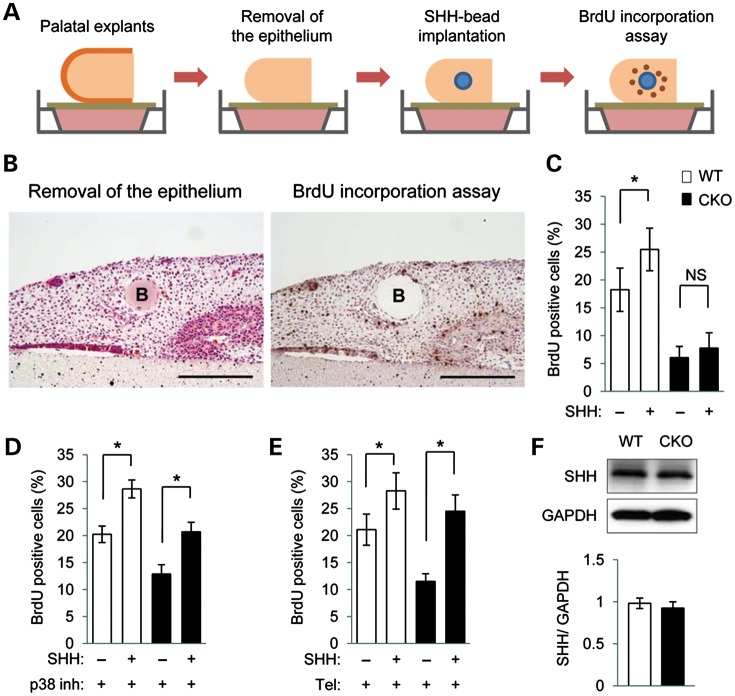
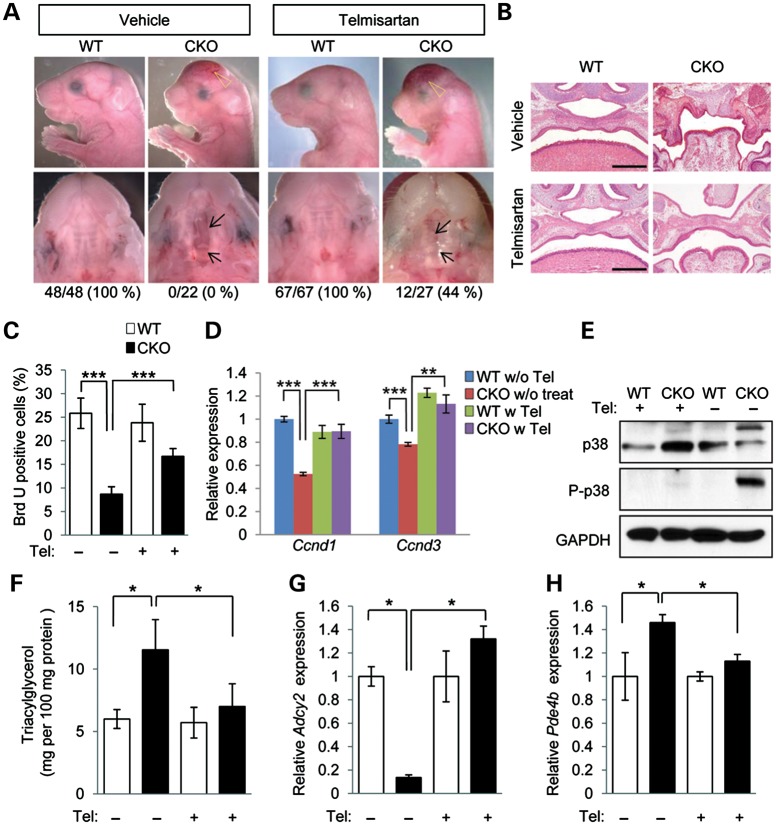
Mutations in transforming growth factor beta (TGFβ) receptor type II (TGFBR2) cause Loeys-Dietz syndrome, characterized by craniofacial and cardiovascular abnormalities. Mice with a deletion of Tgfbr2 in cranial neural crest cells (Tgfbr2(fl/fl);Wnt1-Cre mice) develop cleft palate as the result of abnormal TGFβ signaling activation. However, little is known about metabolic processes downstream of TGFβ signaling during palatogenesis. Here, we show that Tgfbr2 mutant palatal mesenchymal cells spontaneously accumulate lipid droplets, resulting from reduced lipolysis activity. Tgfbr2 mutant palatal mesenchymal cells failed to respond to the cell proliferation stimulator sonic hedgehog, derived from the palatal epithelium. Treatment with p38 mitogen-activated protein kinase (MAPK) inhibitor or telmisartan, a modulator of p38 MAPK activation and lipid metabolism, blocked abnormal TGFβ-mediated p38 MAPK activation, restoring lipid metabolism and cell proliferation activity both in vitro and in vivo. Our results highlight the influence of alternative TGFβ signaling on lipid metabolic activities, as well as how lipid metabolic defects can affect cell proliferation and adversely impact palatogenesis. This discovery has broader implications for the understanding of metabolic defects and potential prevention of congenital birth defects.
Figures
References
-
- Mossey P.A., Little J., Munger R.G., Dixon M.J., Shaw W.C. Cleft lip and palate. Lancet. 2009;374:1773–1785. doi:10.1016/S0140-6736(09)60695-4. - DOI - PubMed
-
- Hrubec T.C., Prater M.R., Toops K.A., Holladay S.D. Reduction in diabetes-induced craniofacial defects by maternal immune stimulation. Birth Defects Res. B Dev. Reprod. Toxicol. 2006;77:1–9. doi:10.1002/bdrb.20062. - DOI - PMC - PubMed
-
- Al Ghafli M.H., Padmanabhan R., Kataya H.H., Berg B. Effects of alpha-lipoic acid supplementation on maternal diabetes-induced growth retardation and congenital anomalies in rat fetuses. Mol. Cell. Biochem. 2004;261:123–135. doi:10.1023/B:MCBI.0000028747.92084.42. - DOI - PubMed
-
- Moore L.L., Singer M.R., Bradlee M.L., Rothman K.J., Milunsky A. A prospective study of the risk of congenital defects associated with maternal obesity and diabetes mellitus. Epidemiology. 2000;11:689–694. doi:10.1097/00001648-200011000-00013. - DOI - PubMed
-
- Ewart-Toland A., Yankowitz J., Winder A., Imagire R., Cox V.A., Aylsworth A.S., Golabi M. Oculoauriculovertebral abnormalities in children of diabetic mothers. Am. J. Med. Genet. 2000;90:303–309. doi:10.1002/(SICI)1096-8628(20000214)90:4<303::AID-AJMG8>3.0.CO;2-Q. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
