

A model for the molecular underpinnings of tooth defects in Axenfeld-Rieger syndrome
- PMID: 23975681
- PMCID: PMC3857954
- DOI: 10.1093/hmg/ddt411
A model for the molecular underpinnings of tooth defects in Axenfeld-Rieger syndrome
Abstract
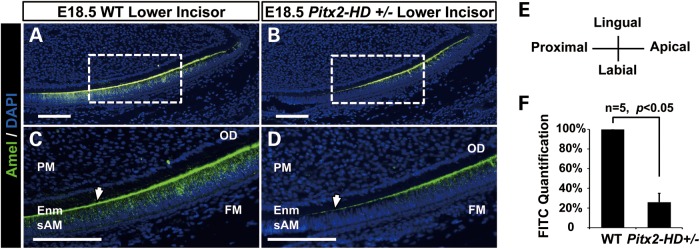
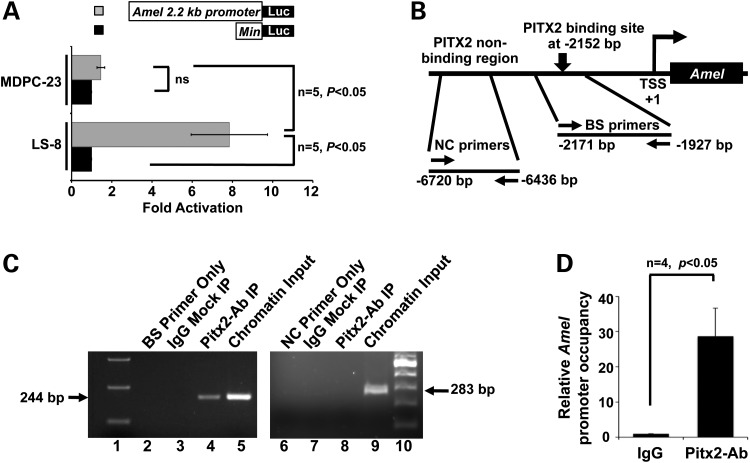
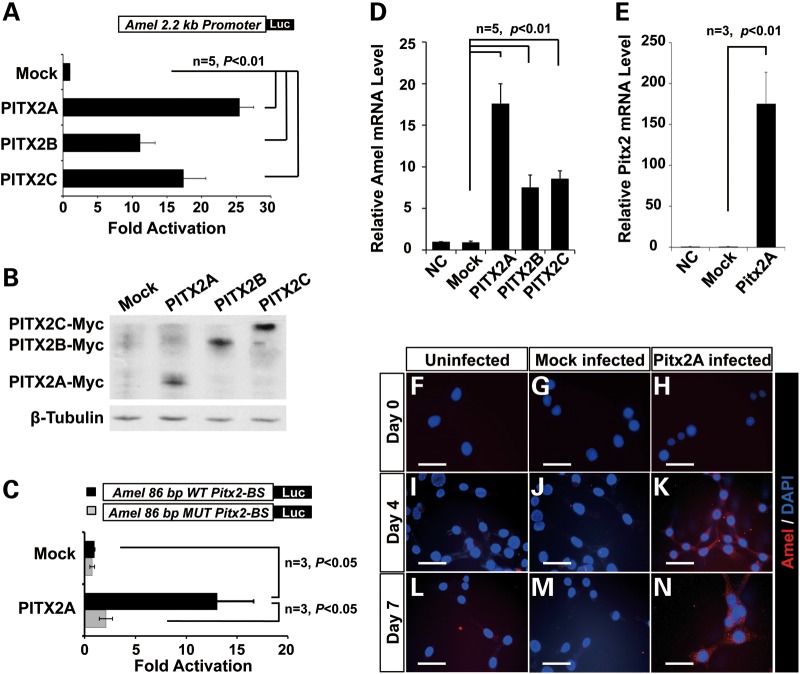
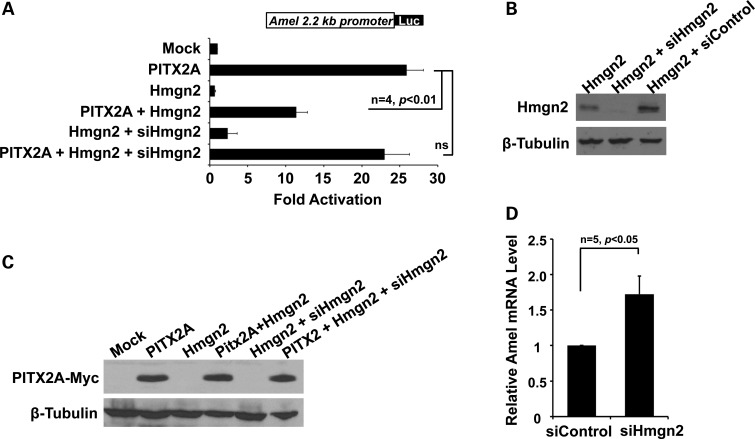
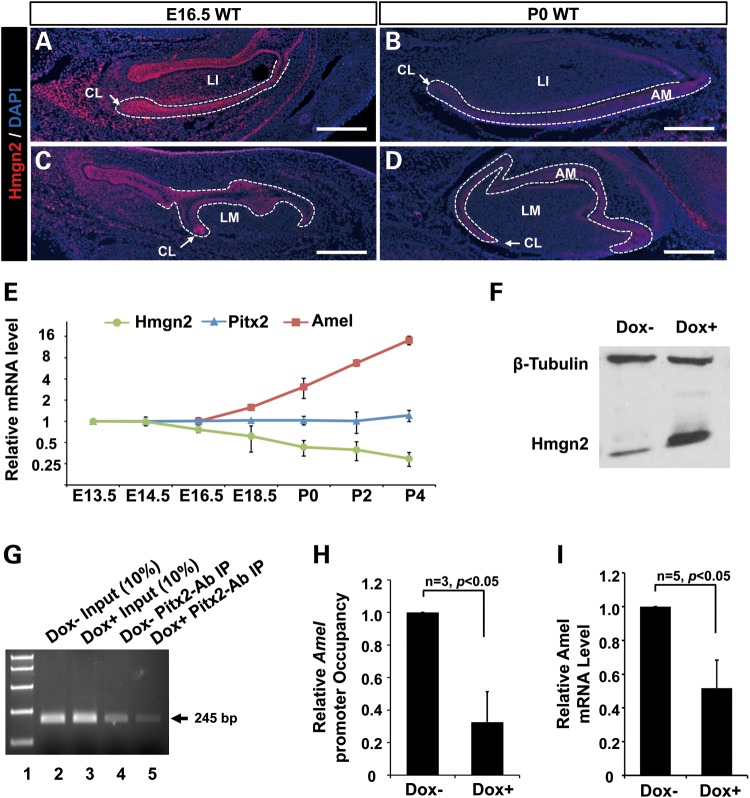
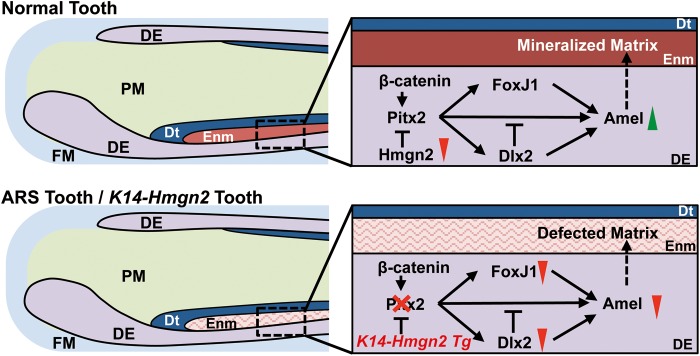
Patients with Axenfeld-Rieger Syndrome (ARS) present various dental abnormalities, including hypodontia, and enamel hypoplasia. ARS is genetically associated with mutations in the PITX2 gene, which encodes one of the earliest transcription factors to initiate tooth development. Thus, Pitx2 has long been considered as an upstream regulator of the transcriptional hierarchy in early tooth development. However, because Pitx2 is also a major regulator of later stages of tooth development, especially during amelogenesis, it is unclear how mutant forms cause ARS dental anomalies. In this report, we outline the transcriptional mechanism that is defective in ARS. We demonstrate that during normal tooth development Pitx2 activates Amelogenin (Amel) expression, whose product is required for enamel formation, and that this regulation is perturbed by missense PITX2 mutations found in ARS patients. We further show that Pitx2-mediated Amel activation is controlled by chromatin-associated factor Hmgn2, and that Hmgn2 prevents Pitx2 from efficiently binding to and activating the Amel promoter. Consistent with a physiological significance to this interaction, we show that K14-Hmgn2 transgenic mice display a severe loss of Amel expression on the labial side of the lower incisors, as well as enamel hypoplasia-consistent with the human ARS phenotype. Collectively, these findings define transcriptional mechanisms involved in normal tooth development and shed light on the molecular underpinnings of the enamel defect observed in ARS patients who carry PITX2 mutations. Moreover, our findings validate the etiology of the enamel defect in a novel mouse model of ARS.
Figures



References
-
- Shields M.B., Buckley E., Klintworth G.K., Thresher R. Axenfeld–Rieger syndrome. A spectrum of developmental disorders. Survey Ophthal. 1985;29:387–409. doi:10.1016/0039-6257(85)90205-X. - DOI - PubMed
-
- Amendt B.A. The Molecular Mechanisms of Axenfeld–Rieger Syndrome. Springer, New York, NY, USA; 2005.
-
- Amendt B., Semina E., Alward W. Rieger syndrome: a clinical, molecular, and biochemical analysis. Cell Mol. Life Sci. 2000;57:1652–1666. doi:10.1007/PL00000647. - DOI - PMC - PubMed
-
- Lines M.A., Kozlowski K., Walter M.A. Molecular genetics of Axenfeld–Rieger malformations. Hum. Mol. Genet. 2002;11:1177–1187. doi:10.1093/hmg/11.10.1177. - DOI - PubMed
-
- Fitch N., Kaback M. The Axenfeld syndrome and the Rieger syndrome. J. Med. Genet. 1978;15:30–34. doi:10.1136/jmg.15.1.30. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
