

Recessive truncating titin gene, TTN, mutations presenting as centronuclear myopathy
- PMID: 23975875
- PMCID: PMC3795603
- DOI: 10.1212/WNL.0b013e3182a6ca62
Recessive truncating titin gene, TTN, mutations presenting as centronuclear myopathy
Abstract
Objective: To identify causative genes for centronuclear myopathies (CNM), a heterogeneous group of rare inherited muscle disorders that often present in infancy or early life with weakness and hypotonia, using next-generation sequencing of whole exomes and genomes.
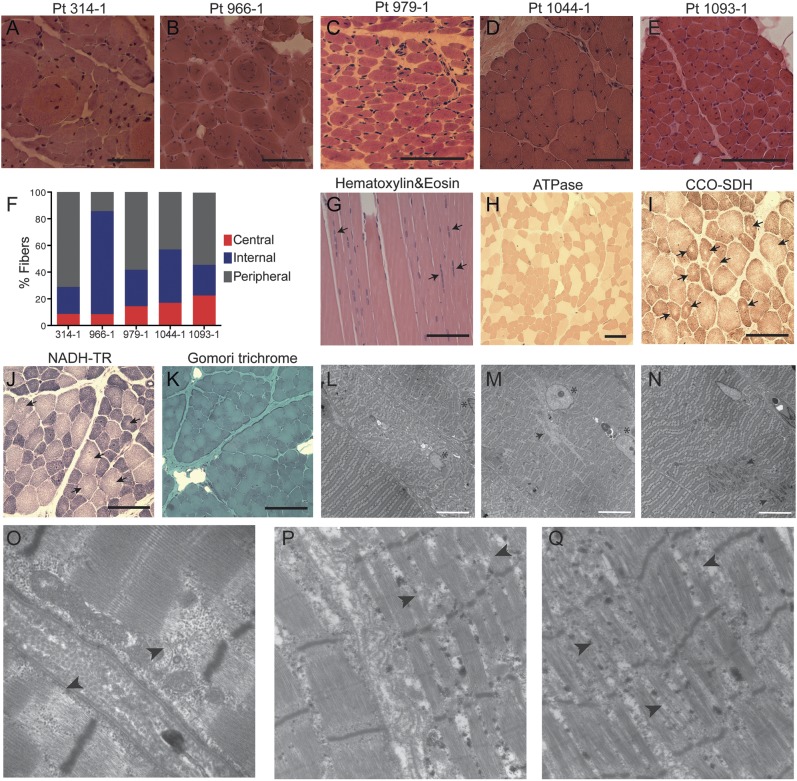
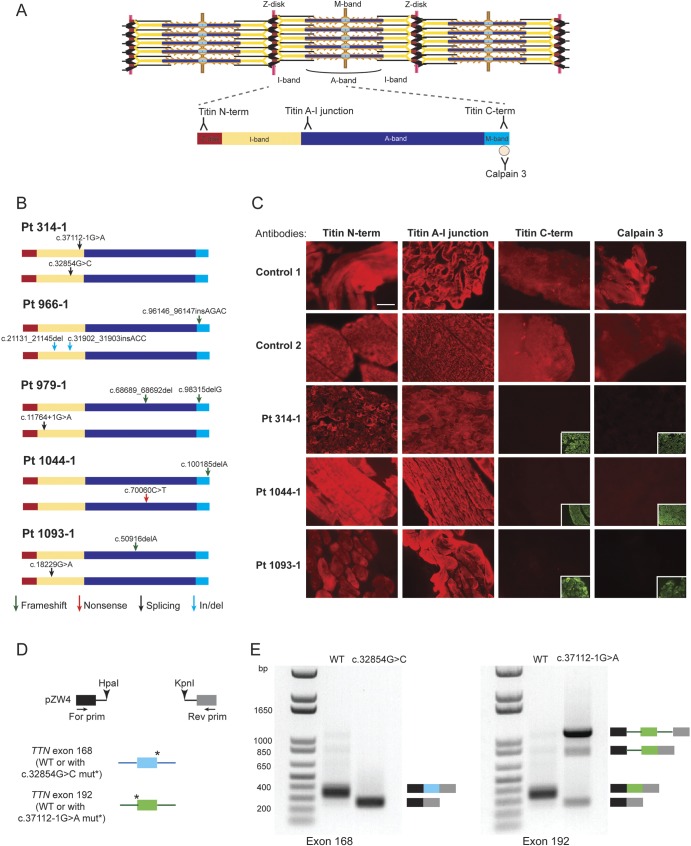
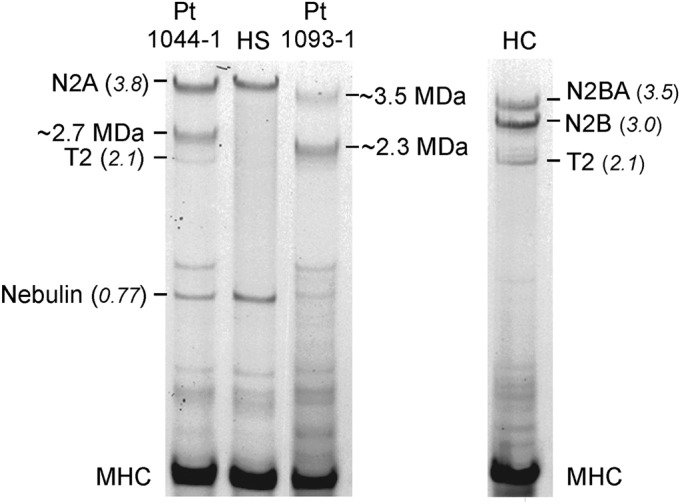
Methods: Whole-exome or -genome sequencing was performed in a cohort of 29 unrelated patients with clinicopathologic diagnoses of CNM or related myopathy depleted for cases with mutations of MTM1, DNM2, and BIN1. Immunofluorescence analyses on muscle biopsies, splicing assays, and gel electrophoresis of patient muscle proteins were performed to determine the molecular consequences of mutations of interest.
Results: Autosomal recessive compound heterozygous truncating mutations of the titin gene, TTN, were identified in 5 individuals. Biochemical analyses demonstrated increased titin degradation and truncated titin proteins in patient muscles, establishing the impact of the mutations.
Conclusions: Our study identifies truncating TTN mutations as a cause of congenital myopathy that is reported as CNM. Unlike the classic CNM genes that are all involved in excitation-contraction coupling at the triad, TTN encodes the giant sarcomeric protein titin, which forms a myofibrillar backbone for the components of the contractile machinery. This study expands the phenotypic spectrum associated with TTN mutations and indicates that TTN mutation analysis should be considered in cases of possible CNM without mutations in the classic CNM genes.
Figures
Comment in
-
Titin and centronuclear myopathy: The tip of the iceberg for TTN-ic mutations?Neurology. 2013 Oct 1;81(14):1189-90. doi: 10.1212/WNL.0b013e3182a6cc43. Epub 2013 Aug 23. Neurology. 2013. PMID: 23975873 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials