

Down-regulation of the oncogene PTTG1 via the KLF6 tumor suppressor during induction of myeloid differentiation
- PMID: 23977008
- PMCID: PMC3745464
- DOI: 10.1371/journal.pone.0071282
Down-regulation of the oncogene PTTG1 via the KLF6 tumor suppressor during induction of myeloid differentiation
Abstract
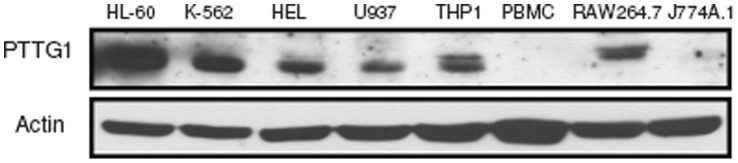
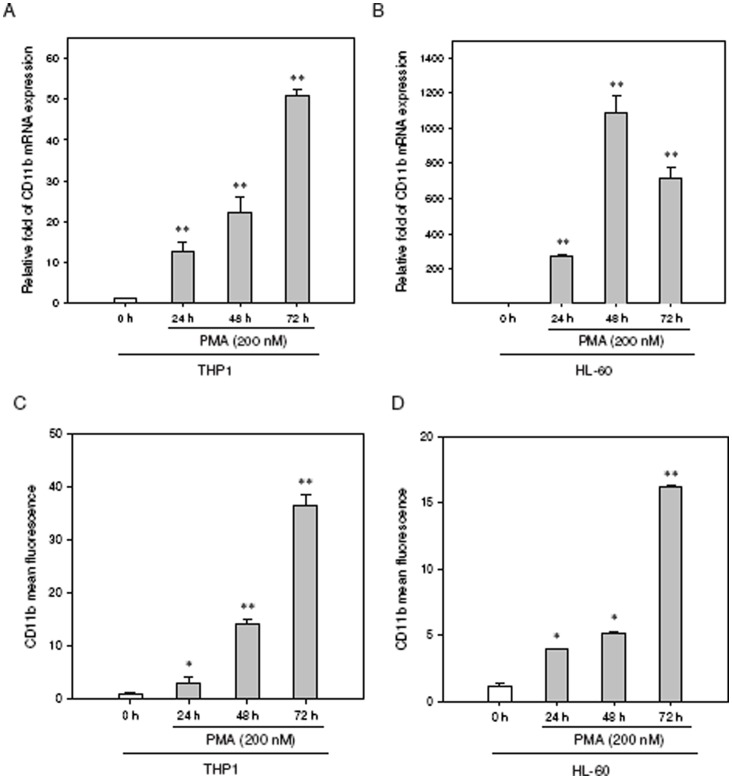
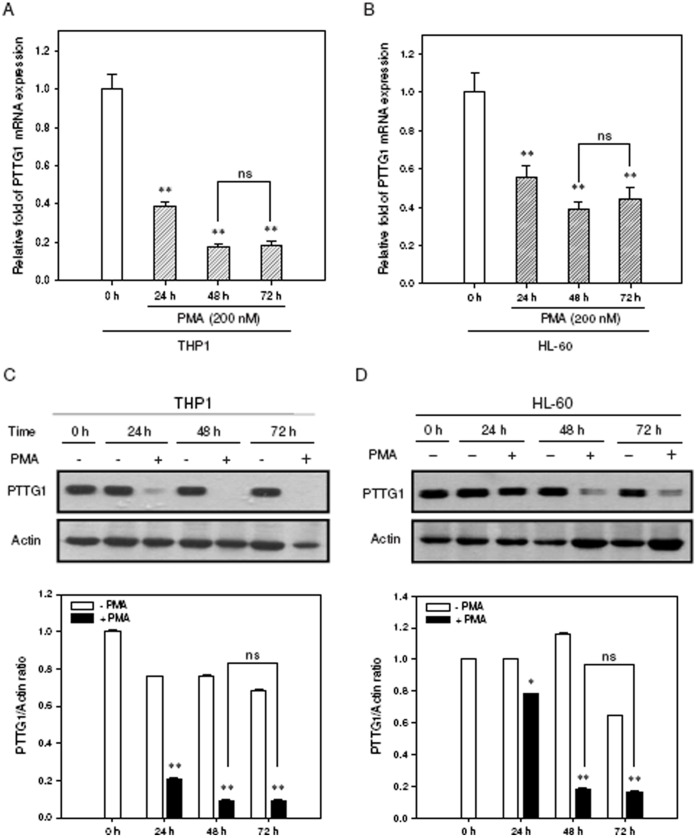
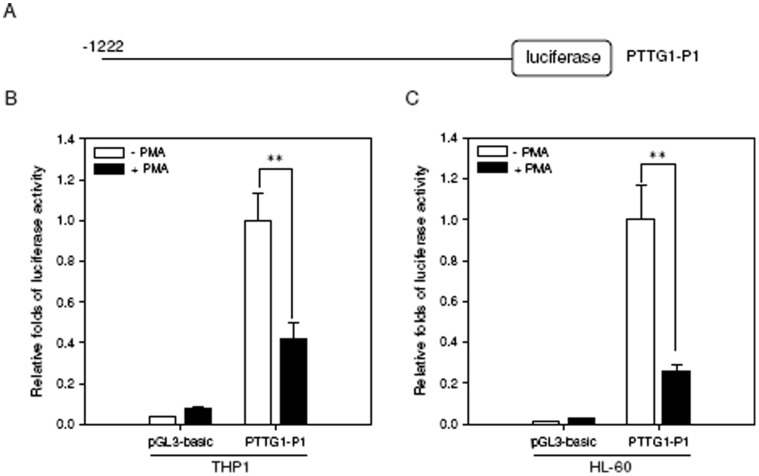
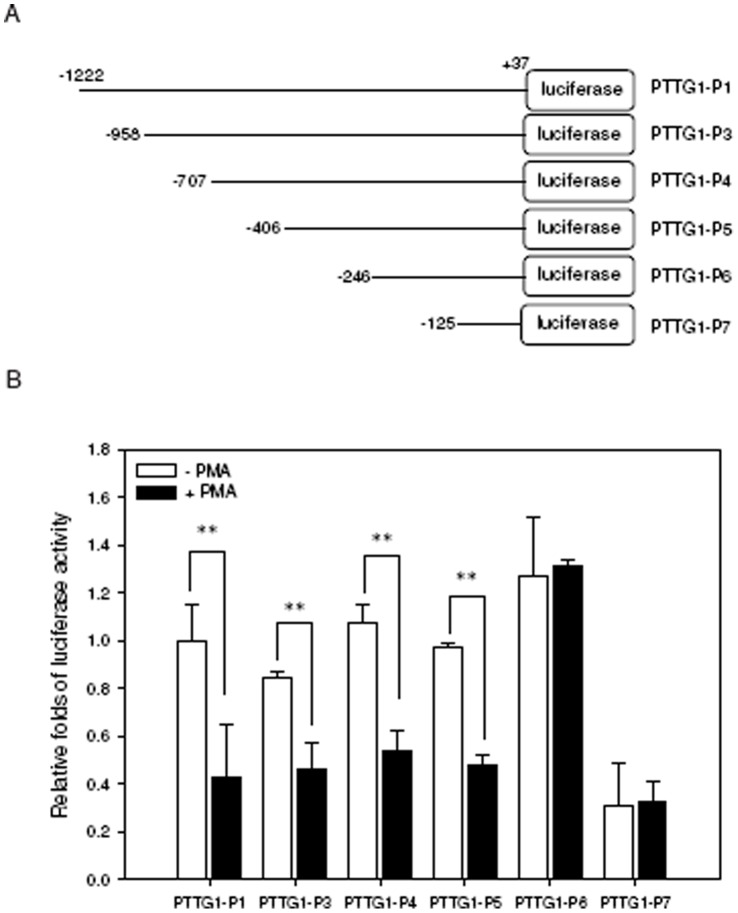
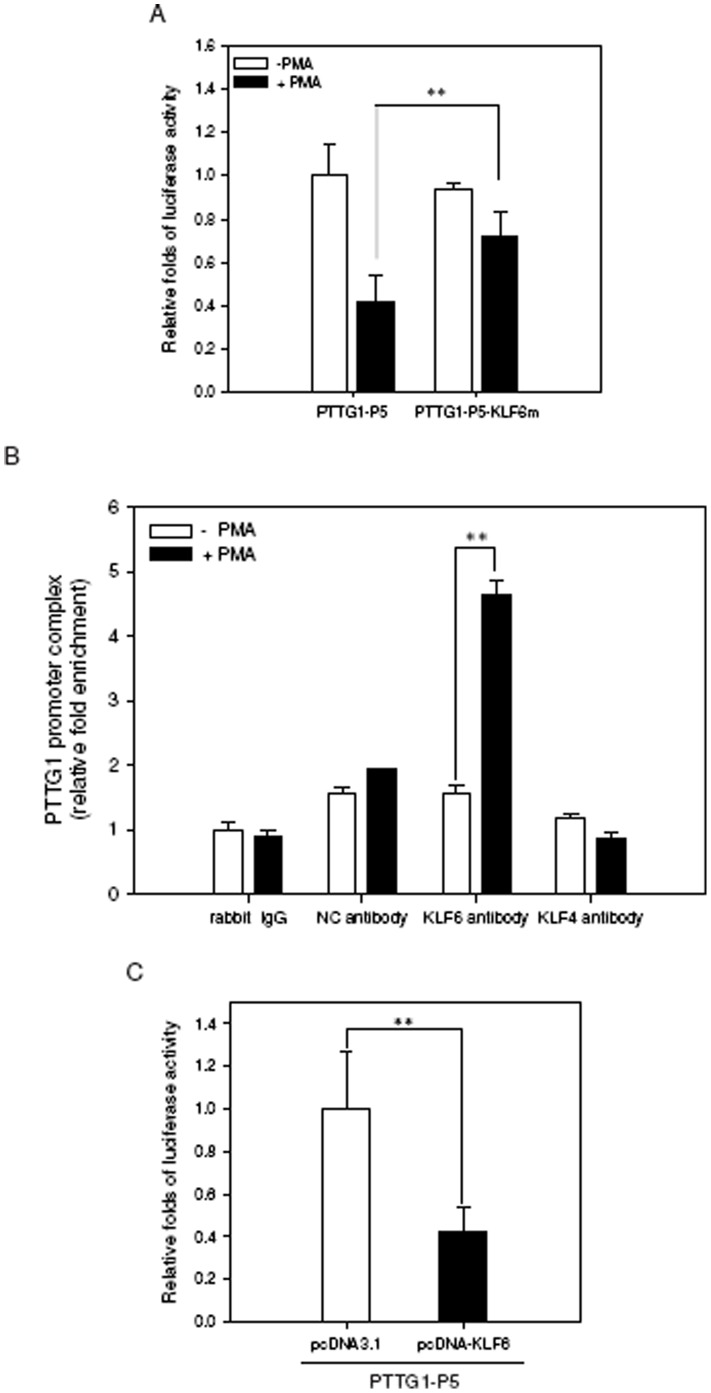
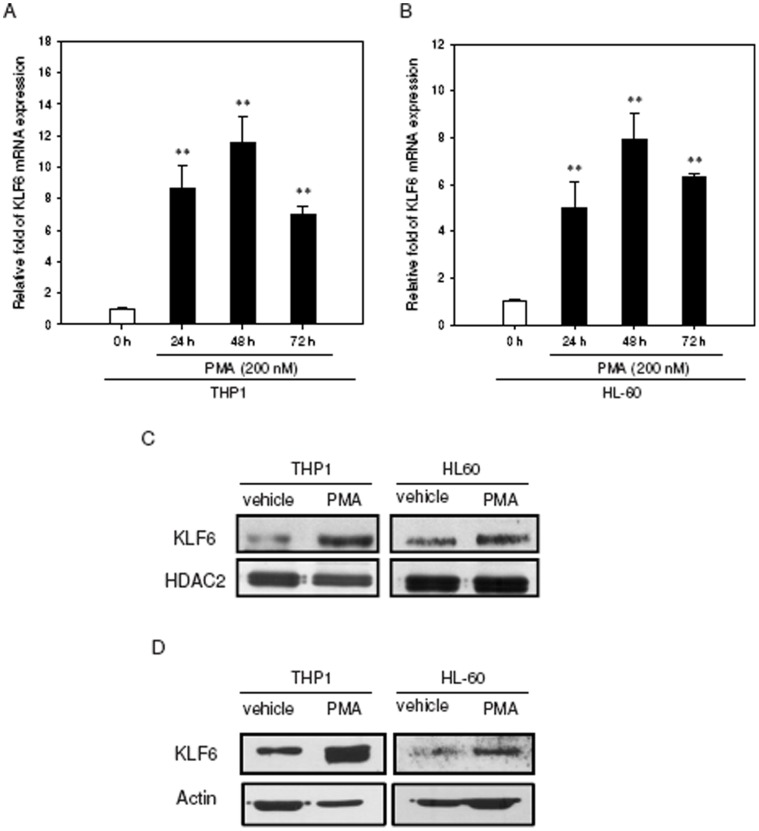
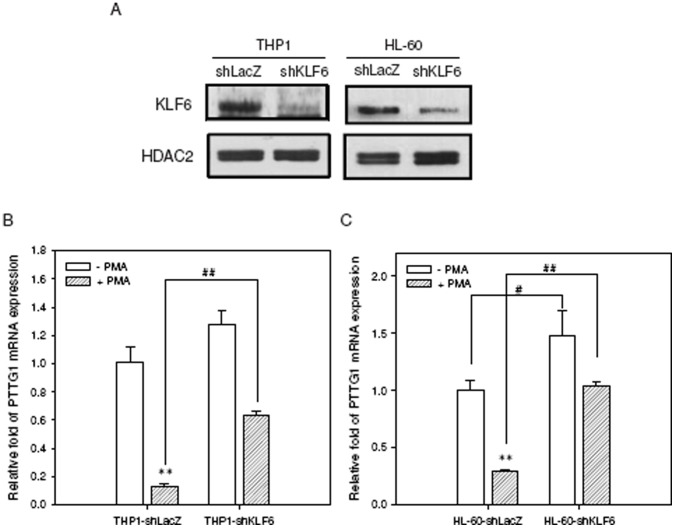
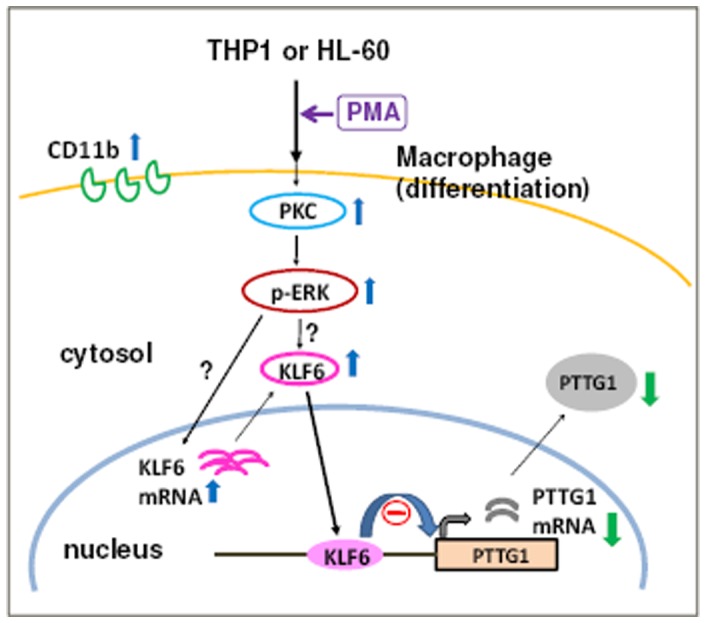
The aberrant expression of proto-oncogenes is involved in processes that are responsible for cellular proliferation and the inhibition of myeloid differentiation in acute myeloid leukemia (AML). Pituitary Tumor-Transforming gene 1 (PTTG1), an oncogenic transcription factor, is abundantly expressed in various human cancers and hematopoietic malignancies. However, its expression in normal leukocytes and most normal tissues is very low or undetectable. The mechanism by which PTTG1 overexpression modifies myeloid cell development and promotes leukemogenesis remain unclear. To investigate the mechanistic links between PTTG1 overexpression and leukemia cell differentiation, we utilized phorbol 12-myristate 13-acetate (PMA), a well-known agent that triggers monocyte/macrophage differentiation, to analyze the expression patterns of PTTG1 in PMA-induced myeloid differentiation. We found that PTTG1 is down-regulated at the transcriptional level in PMA-treated HL-60 and THP1 cells. In addition, we identified a binding site for a tumor suppressor protein, Kruppel-like factor 6 (KLF6), in the PTTG1 promoter. We found that KLF6 could directly bind and repress PTTG1 expression. In HL-60 and THP1 cells, KLF6 mRNA and protein levels are up-regulated with a concordant reduction of PTTG1 expression upon treatment with PMA. Furthermore, KLF6 knockdown by shRNA abolished the suppression of PTTG1 and reduced the activation of the differentiation marker CD11b in PMA-primed cells. The protein kinase C (PKC) inhibitor and the MAPK/ERK kinase (MEK) inhibitor significantly blocked the potentiation of PMA-mediated KLF6 induction and the down-regulation of PTTG1, indicating that PTTG1 is suppressed via the activation of PKC/ERK/KLF6 pathway. Our findings suggest that drugs that increase the KLF6 inhibition of PTTG1 may have a therapeutic application in AML treatment strategies.
Conflict of interest statement
Figures

Similar articles
-
Tumor suppressor activity of KLF6 mediated by downregulation of the PTTG1 oncogene.FEBS Lett. 2010 Mar 5;584(5):1006-10. doi: 10.1016/j.febslet.2010.01.049. Epub 2010 Jan 30. FEBS Lett. 2010. PMID: 20116377 Free PMC article.
-
Activation of protein kinase C by phorbol 12-myristate 13-acetate suppresses the growth of lung cancer cells through KLF6 induction.Cancer Biol Ther. 2009 May;8(9):801-7. doi: 10.4161/cbt.8.9.8186. Epub 2009 May 17. Cancer Biol Ther. 2009. PMID: 19333010
-
Knockdown of KLF6 ameliorates myocardial infarction by regulating autophagy via transcriptional regulation of PTTG1.Am J Physiol Cell Physiol. 2025 Jan 1;328(1):C115-C127. doi: 10.1152/ajpcell.00191.2024. Epub 2024 Dec 9. Am J Physiol Cell Physiol. 2025. PMID: 39652418
-
Biology of Krüppel-like factor 6 transcriptional regulator in cell life and death.IUBMB Life. 2010 Dec;62(12):896-905. doi: 10.1002/iub.396. IUBMB Life. 2010. PMID: 21154818 Review.
-
The role of KLF6 and its splice variants in cancer therapy.Drug Resist Updat. 2009 Feb-Apr;12(1-2):1-7. doi: 10.1016/j.drup.2008.11.001. Epub 2008 Dec 18. Drug Resist Updat. 2009. PMID: 19097929 Review.
Cited by
-
Angiostrongylus cantonensis Galectin-1 interacts with Annexin A2 to impair the viability of macrophages via activating JNK pathway.Parasit Vectors. 2020 Apr 8;13(1):183. doi: 10.1186/s13071-020-04038-w. Parasit Vectors. 2020. PMID: 32268913 Free PMC article.
-
Macrophage maturation from blood monocytes is altered in people with HIV, and is linked to serum lipid profiles and activation indices: A model for studying atherogenic mechanisms.PLoS Pathog. 2020 Oct 1;16(10):e1008869. doi: 10.1371/journal.ppat.1008869. eCollection 2020 Oct. PLoS Pathog. 2020. PMID: 33002093 Free PMC article.
-
Pituitary Tumor-Transforming Gene 1/Delta like Non-Canonical Notch Ligand 1 Signaling in Chronic Liver Diseases.Int J Mol Sci. 2022 Jun 21;23(13):6897. doi: 10.3390/ijms23136897. Int J Mol Sci. 2022. PMID: 35805898 Free PMC article. Review.
-
Response of Myeloid Leukemia Cells to Luteolin is Modulated by Differentially Expressed Pituitary Tumor-Transforming Gene 1 (PTTG1) Oncoprotein.Int J Mol Sci. 2018 Apr 12;19(4):1173. doi: 10.3390/ijms19041173. Int J Mol Sci. 2018. PMID: 29649138 Free PMC article.
-
PTTG1 promotes M2 macrophage polarization via the cGMP-PKG signaling pathway and facilitates EMT progression in human epithelial ovarian cancer cells.Discov Oncol. 2025 May 12;16(1):730. doi: 10.1007/s12672-025-02512-4. Discov Oncol. 2025. PMID: 40353994 Free PMC article.
References
-
- Crans-Vargas HN, Landaw EM, Bhatia S, Sandusky G, Moore TB, et al. (2002) Expression of cyclic adenosine monophosphate response-element binding protein in acute leukemia. Blood 99: 2617–2619. - PubMed
-
- Shima Y, Kitabayashi I (2011) Deregulated transcription factors in leukemia. Int J Hematol 94: 134–141. - PubMed
-
- Pei L, Melmed S (1997) Isolation and characterization of a pituitary tumor-transforming gene (PTTG). Mol Endocrinol 11: 433–441. - PubMed
-
- Dominguez A, Ramos-Morales F, Romero F, Rios RM, Dreyfus F, et al. (1998) hpttg, a human homologue of rat pttg, is overexpressed in hematopoietic neoplasms. Evidence for a transcriptional activation function of hPTTG. Oncogene 17: 2187–2193. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
