

Molecular dynamics simulations to provide insights into epitopes coupled to the soluble and membrane-bound MHC-II complexes
- PMID: 23977319
- PMCID: PMC3747130
- DOI: 10.1371/journal.pone.0072575
Molecular dynamics simulations to provide insights into epitopes coupled to the soluble and membrane-bound MHC-II complexes
Abstract
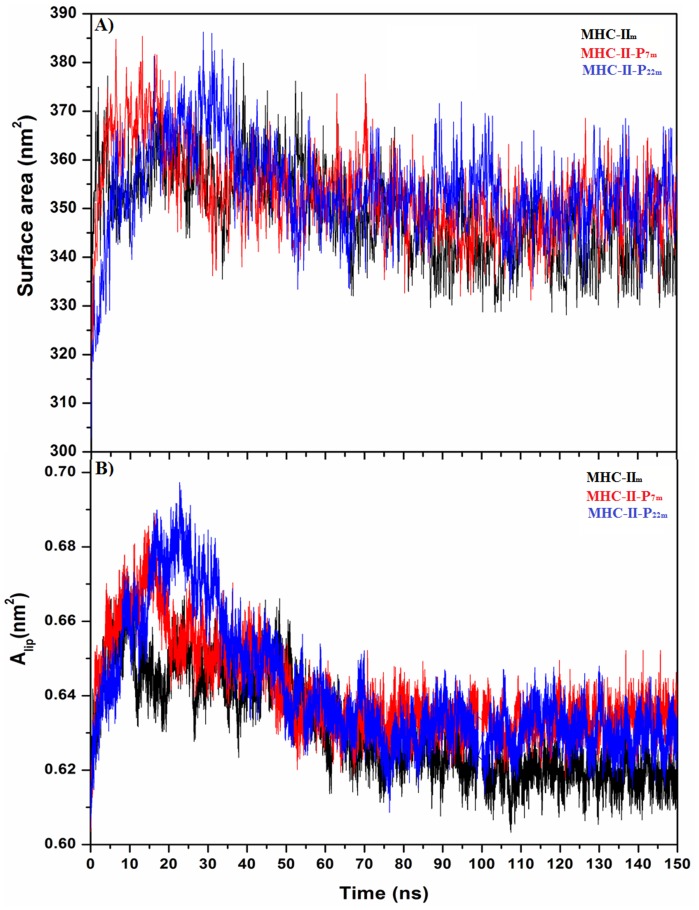
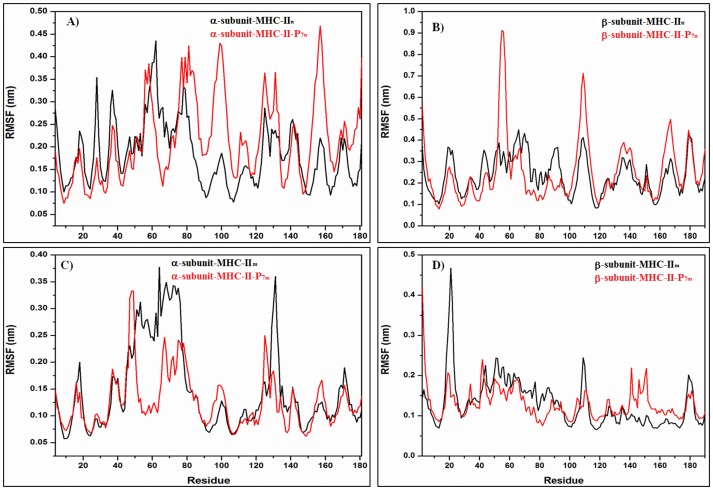
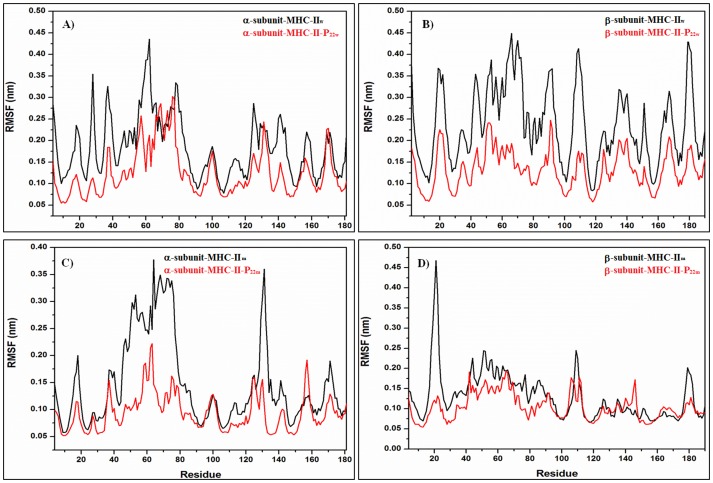
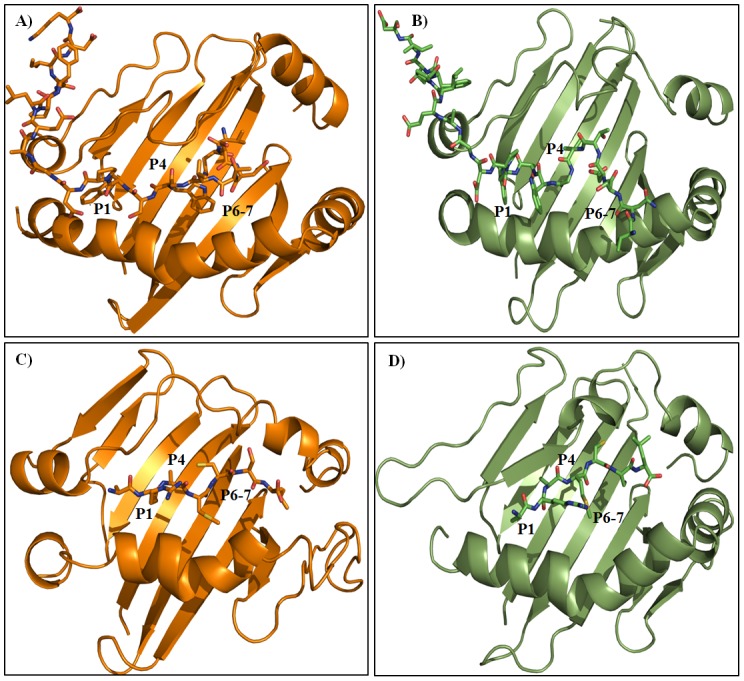
Epitope recognition by major histocompatibility complex II (MHC-II) is essential for the activation of immunological responses to infectious diseases. Several studies have demonstrated that this molecular event takes place in the MHC-II peptide-binding groove constituted by the α and β light chains of the heterodimer. This MHC-II peptide-binding groove has several pockets (P1-P11) involved in peptide recognition and complex stabilization that have been probed through crystallographic experiments and in silico calculations. However, most of these theoretical calculations have been performed without taking into consideration the heavy chains, which could generate misleading information about conformational mobility both in water and in the membrane environment. Therefore, in absence of structural information about the difference in the conformational changes between the peptide-free and peptide-bound states (pMHC-II) when the system is soluble in an aqueous environment or non-covalently bound to a cell membrane, as the physiological environment for MHC-II is. In this study, we explored the mechanistic basis of these MHC-II components using molecular dynamics (MD) simulations in which MHC-II was previously co-crystallized with a small epitope (P7) or coupled by docking procedures to a large (P22) epitope. These MD simulations were performed at 310 K over 100 ns for the water-soluble (MHC-IIw, MHC-II-P(7w), and MHC-II-P(22w)) and 150 ns for the membrane-bound species (MHC-IIm, MHC-II-P(7m), and MHC-II-P(22m)). Our results reveal that despite the different epitope sizes and MD simulation environments, both peptides are stabilized primarily by residues lining P1, P4, and P6-7, and similar noncovalent intermolecular energies were observed for the soluble and membrane-bound complexes. However, there were remarkably differences in the conformational mobility and intramolecular energies upon complex formation, causing some differences with respect to how the two peptides are stabilized in the peptide-binding groove.
Conflict of interest statement
Figures

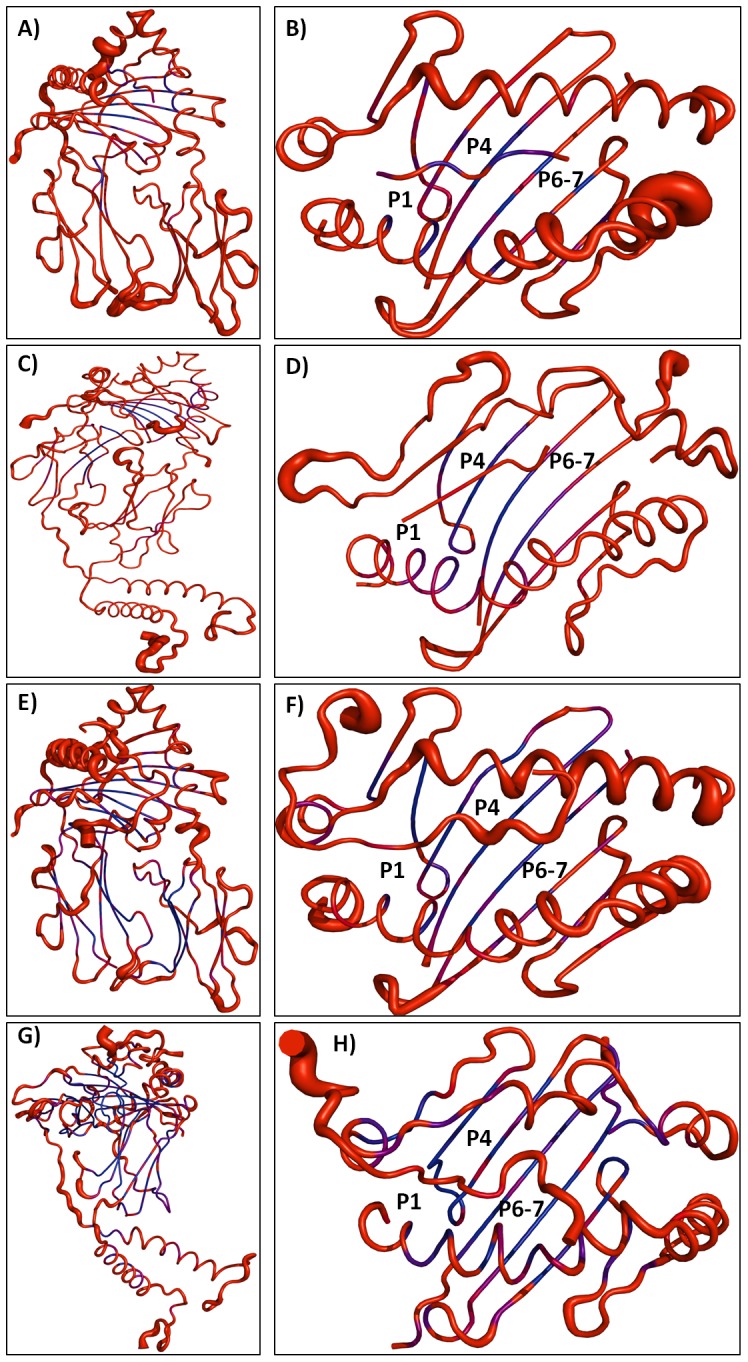
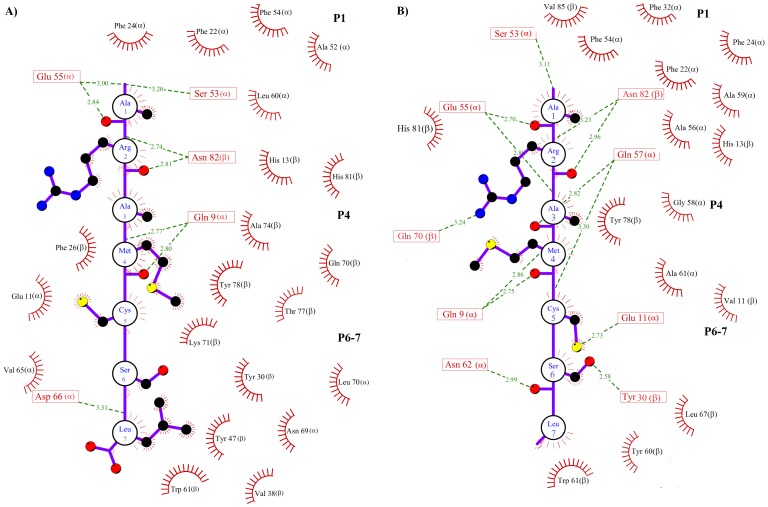
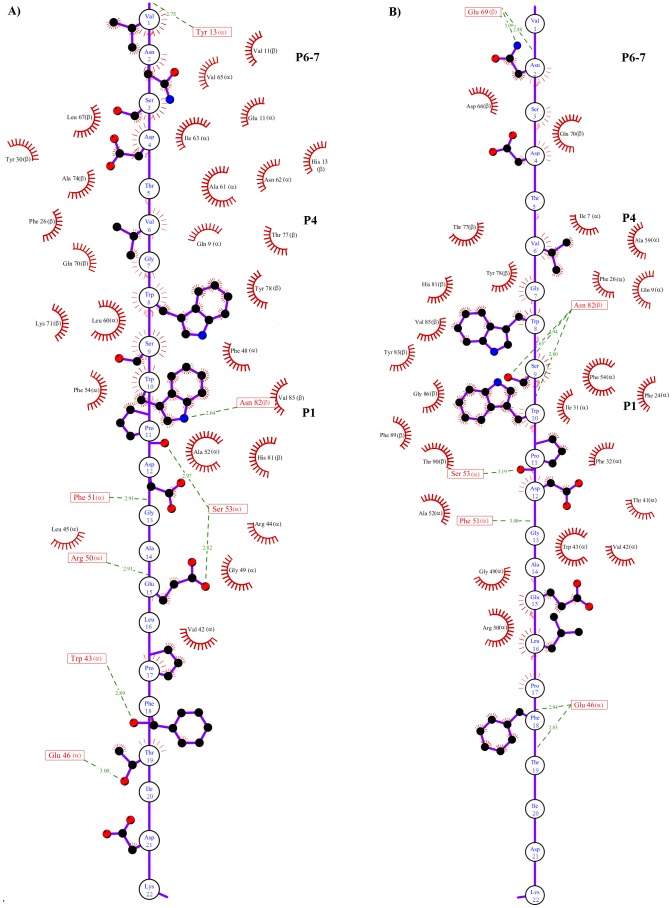
References
-
- Trombetta ES, Mellman I (2005) Cell biology of antigen processing in vitro and in vivo. Annu Rev Immunol 23: 975–1028. - PubMed
-
- Rammensee HG (1995) Chemistry of peptides associated with MHCclass I and class II molecules. Curr Opin Immunol 7: 85–96. - PubMed
-
- Rudensky AY, Preston-Hurlburt P, Hong SC, Barlow A, Janeway CA (1991) Sequence analysis of peptides bound to MHC class II molecules. Nature 353: 622–627. - PubMed
-
- Suri A, Lovitch SB, Unanue ER (2006) The wide diversity and complexity of peptides bound to class II MHC molecules. Curr Opin Immunol 18: 70–77. - PubMed
-
- Stern LJ, Wiley DC (1994) Antigenic peptide binding by class I and class II histocompatibility proteins. Structure 2: 245–251. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
