

Review
doi: 10.1038/nrmicro3093.
Epub 2013 Sep 2.
MLST revisited: the gene-by-gene approach to bacterial genomics
Affiliations
- PMID: 23979428
- PMCID: PMC3980634
- DOI: 10.1038/nrmicro3093
Item in Clipboard
Review
MLST revisited: the gene-by-gene approach to bacterial genomics
Nat Rev Microbiol.
2013 Oct.
Abstract
Multilocus sequence typing (MLST) was proposed in 1998 as a portable sequence-based method for identifying clonal relationships among bacteria. Today, in the whole-genome era of microbiology, the need for systematic, standardized descriptions of bacterial genotypic variation remains a priority. Here, to meet this need, we draw on the successes of MLST and 16S rRNA gene sequencing to propose a hierarchical gene-by-gene approach that reflects functional and evolutionary relationships and catalogues bacteria 'from domain to strain'. Our gene-based typing approach using online platforms such as the Bacterial Isolate Genome Sequence Database (BIGSdb) allows the scalable organization and analysis of whole-genome sequence data.
Figures
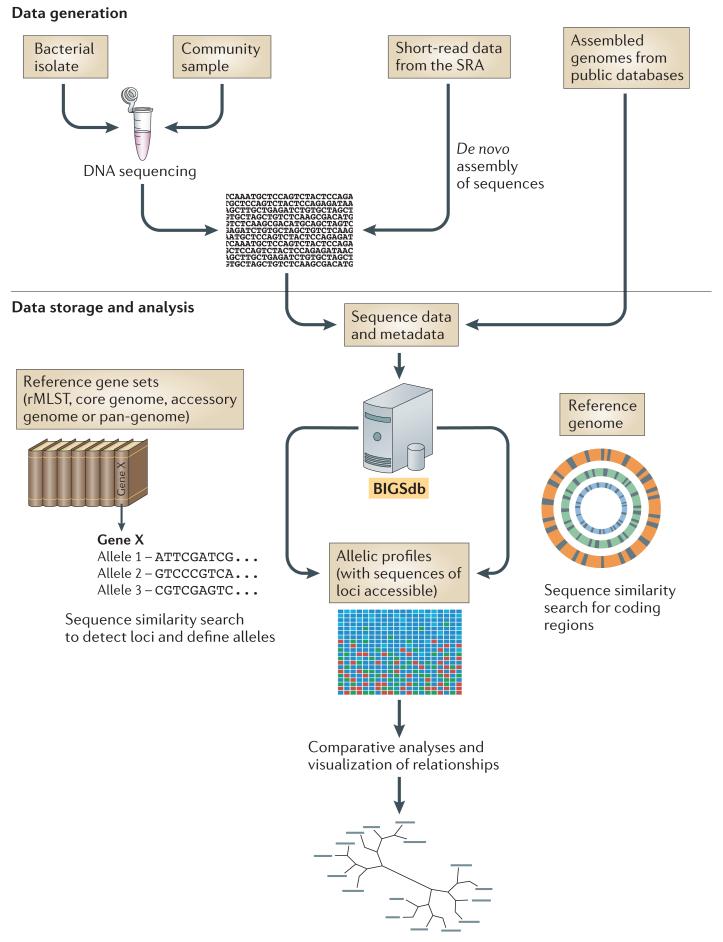
The gene-by-gene approach can be used to integrate whole-genome sequencing (WGS) data with isolate data, facilitating easy storage and retrieval for downstream analyses. WGS data can be obtained in several ways: DNA can be isolated from a bacterial isolate or community sample and sequenced on an appropriate platform, short-read data can be obtained from the Sequence Read Archive (SRA), and assembled genomes can be downloaded from public databases (for example, GenBank). In the cases of sequencing and SRA data, short-read data are assembled with an appropriate algorithm, directed by the sequencing platform used. Assembled contigs are uploaded to a Bacterial Isolate Genome Sequence Database (BIGSdb), and they can then be compared against either sets of reference genes or a reference genome using algorithms such as BLAST. Reference gene sets can be tailored to meet particular requirements and thus range from collections of loci that are useful for epidemiological investigations to subsets of genes with functional relevance, for example in a metabolic pathway. For comparisons based on both reference gene sets and reference genomes, the nucleotide sequences remain accessible, but loci are assigned allele designations to generate an allelic profile as for multilocus sequence typing (MLST). The Genome Comparator module in BIGSdb can be used to produce a distance matrix based on allelic profiles, and this matrix can in turn be used to visualize relationships between isolates using an appropriate algorithm, such as NeighbourNet in SplitsTree. Alternatively, downstream analyses of the aligned sequence data can be carried out by exporting sequence data to external packages. In addition to a distance matrix and sequence alignments, the Genome Comparator outputs also include a table showing which loci are identical and which are different among the isolates examined. rMLST, ribosomal MLST.
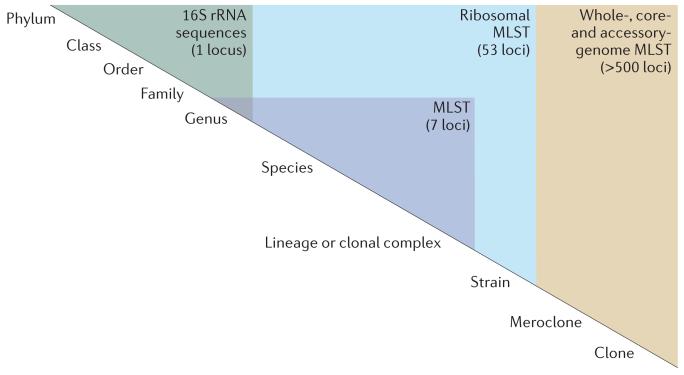
Hierarchical nomenclature schemes are artificial constructs that are developed to facilitate communication and are subject to change as new information becomes available. Nomenclature schemes are dependent on various factors, including sequence relationships, and are ideally genealogically based. The challenge is to map whole-genome sequencing (WGS) data to nomenclature schemes transparently but flexibly at a range of resolutions. The highest discrimination is required for studies of bacterial isolates from one patient or from very closely related transmission chains; these isolates can be thought of as having undergone microevolution. Such studies will require comparisons of whole genomes using whole-genome multilocus sequence typing (MLST). Progressively lower resolution is required for studies of isolates with more distant common ancestors and, therefore, with more genetic differences. These relationships are best studied using the core genome common to the set of isolates of interest. Genes encoding ribosomal proteins are a particularly useful subset of core genes, and ribosomal MLST accommodates many levels of genealogical relationships, from clonal complexes and lineages to species, genera and beyond. In a database such as the Bacterial Isolate Genome Sequence Database (BIGSdb), multiple gene-by-gene schemes can be implemented alongside other, more conventional sequence-based schemes. Particular genotype summaries of genes or collections of genes can be associated with particular nomenclature schemes, enabling the database to deliver a plain-language report to the user.
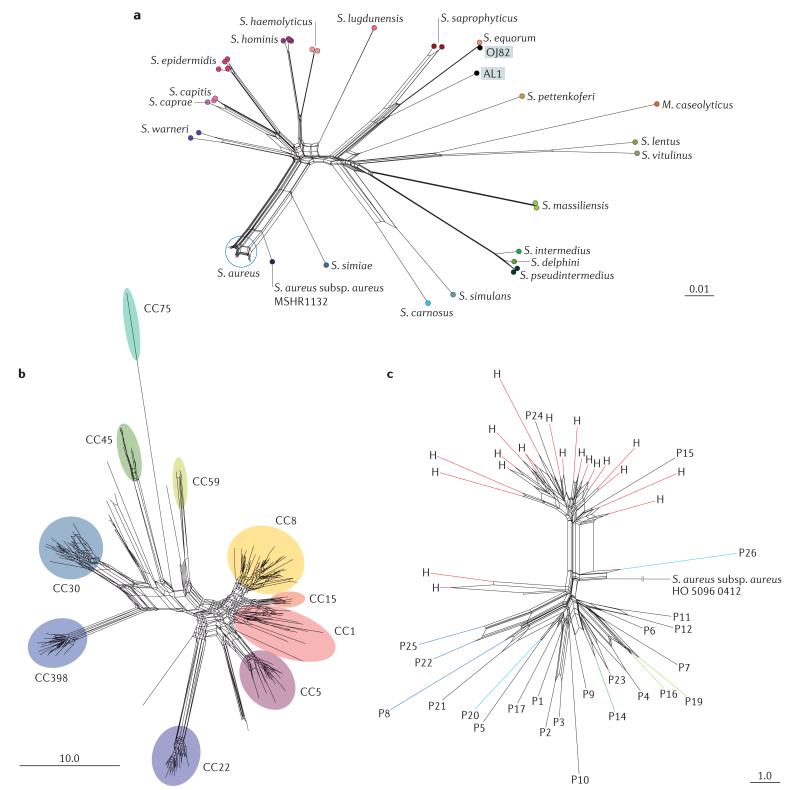
These analyses were carried out using both the Genome Comparator module within the Bacterial Isolate Genome Sequence Database (BIGSdb) platform and data publicly available within the PubMLST website. The phylogenetic networks were generated using the NeighbourNet algorithm in SplitsTree (v4.12.3). a | Resolution of 52 staphylococcal isolates on the basis of nucleotide sequence diversity at 51 ribosomal multilocus sequence typing (rMLST) loci, permitting the determination of the species assignment of two recently described isolates, Staphylococcus sp. OJ82 and Staphylococcus sp. AL1. Staphylococcus sp. OJ82 probably corresponds to Stapylococcus equorum, whereas Staphylococcus sp. AL1 is related to, but distinct from, S. equorum and Staphylococcus saprophyticus. All species shown are in the genus Staphylococcus, except for Macrococcus caseolyticus. b | The diversity of 669 Staphylococcus aureus isolates on the basis of allelic diversity at 51 rMLST loci. The extensive diversity of S. aureus is illustrated here; the rMLST clustering is congruent with MLST clonal complexes (CCs; indicated) and indicates relationships among the isolates. c | Resolution of multidrug-resistant S. aureus (MRSA) isolates from an outbreak in a special-care baby unit, using a gene-by-gene comparison to a reference genome (S. aureus subsp. aureus HO 5096 0412). Twenty isolates obtained from a health care worker are indicated with the letter H and shown in red, whereas patient isolates are indicated with a letter P. Groups of isolates from patients who were members of the same family are shown in the same colour. Reticulations in the diagrams indicate departures from a strictly tree-like phylogeny; this can have a number of causes, including homoplasy as a result of recombination, mutation or lack of resolution. Such graphs are rapidly produced and represent the relationships among sets of genome data; these relationships can then be readily used to resolve isolate relationships in clinical and other settings. Scale bars represent distances calculated from the nucleotide sequence alignment (part a) and number of loci (parts b,c)
References
-
- Ciccarelli FD, et al. Toward automatic reconstruction of a highly resolved tree of life. Science. 2006;311:1283–1287. - PubMed
-
- Medini D, et al. Microbiology in the post-genomic era. Nat. Rev. Microbiol. 2008;6:419–430. - PubMed
-
- Fournier PE, Raoult D. Prospects for the future using genomics and proteomics in clinical microbiology. Annu. Rev. Microbiol. 2011;65:169–188. - PubMed
-
- Stackebrandt E, et al. Report of the ad hoc committee for the re-evaluation of the species definition in bacteriology. Int. J. Syst. Evol. Microbiol. 2002;52:1043–1047. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
