

SUMO and Alzheimer's disease
- PMID: 23979993
- PMCID: PMC3823823
- DOI: 10.1007/s12017-013-8257-7
SUMO and Alzheimer's disease
Abstract
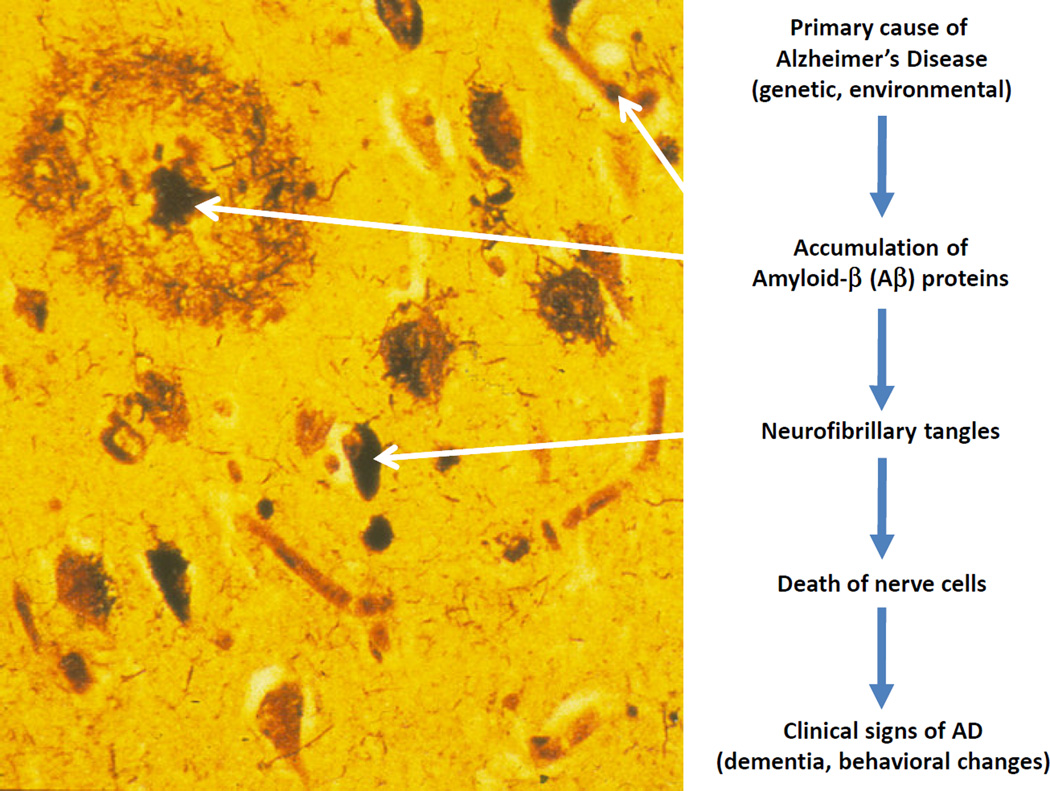
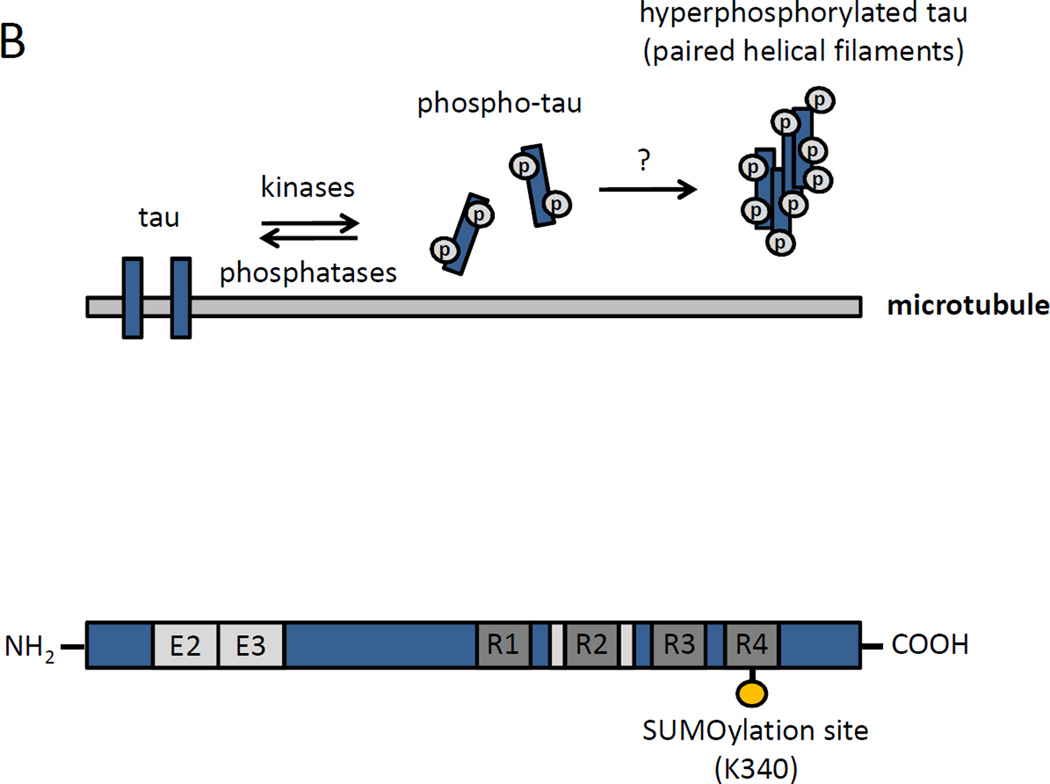
Alzheimer's disease (AD) is a neurodegenerative disorder characterized by progressive cognitive decline and is the most common cause of dementia in the elderly. Histopathologically, AD features insoluble aggregates of two proteins in the brain, amyloid-β (Aβ) and the microtubule-associated protein tau, both of which have been linked to the small ubiquitin-like modifier (SUMO). A large body of research has elucidated many of the molecular and cellular pathways that underlie AD, including those involving the abnormal Aβ and tau aggregates. However, a full understanding of the etiology and pathogenesis of the disease has remained elusive. Consequently, there are currently no effective therapeutic options that can modify the disease progression and slow or stop the decline of cognitive functioning. As part of the effort to address this lacking, there needs a better understanding of the signaling pathways that become impaired under AD pathology, including the regulatory mechanisms that normally control those networks. One such mechanism involves SUMOylation, which is a post-translational modification (PTM) that is involved in regulating many aspects of cell biology and has also been found to have several critical neuron-specific roles. Early studies have indicated that the SUMO system is likely altered with AD-type pathology, which may impact Aβ levels and tau aggregation. Although still a relatively unexplored topic, SUMOylation will likely emerge as a significant factor in AD pathogenesis in ways which may be somewhat analogous to other regulatory PTMs such as phosphorylation. Thus, in addition to the upstream effects on tau and Aβ processing, there may also be downstream effects mediated by Aβ aggregates or other AD-related factors on SUMO-regulated signaling pathways. Multiple proteins that have functions relevant to AD pathology have been identified as SUMO substrates, including those involved in synaptic physiology, mitochondrial dynamics, and inflammatory signaling. Ongoing studies will determine how these SUMO-regulated functions in neurons and glial cells may be impacted by Aβ and AD pathology. Here, we present a review of the current literature on the involvement of SUMO in AD, as well as an overview of the SUMOylated proteins and pathways that are potentially dysregulated with AD pathogenesis.
Figures


References
-
- Abramov E, Dolev I, Fogel H, Ciccotosto GD, Ruff E, Slutsky I. Amyloid-beta as a positive endogenous regulator of release probability at hippocampal synapses. Nat Neurosci. 2009;12(12):1567–1576. - PubMed
-
- Akama KT, Van Eldik LJ. Beta-amyloid stimulation of inducible nitric-oxide synthase in astrocytes is interleukin-1beta- and tumor necrosis factor-alpha (TNFalpha)-dependent, and involves a TNFalpha receptor-associated factor- and NFkappaB-inducing kinase-dependent signaling mechanism. J Biol Chem. 2000;275(11):7918–7924. - PubMed
-
- Alzheimer's A. 2012 Alzheimer's disease facts and figures. Alzheimers Dement. 2012;8(2):131–168. - PubMed
-
- Arendt T. Synaptic degeneration in Alzheimer's disease. Acta Neuropathol. 2009;118(1):167–179. [Review]. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
