

Polo-like kinase 2 regulates selective autophagic α-synuclein clearance and suppresses its toxicity in vivo
- PMID: 23983262
- PMCID: PMC3799334
- DOI: 10.1073/pnas.1309991110
Polo-like kinase 2 regulates selective autophagic α-synuclein clearance and suppresses its toxicity in vivo
Abstract
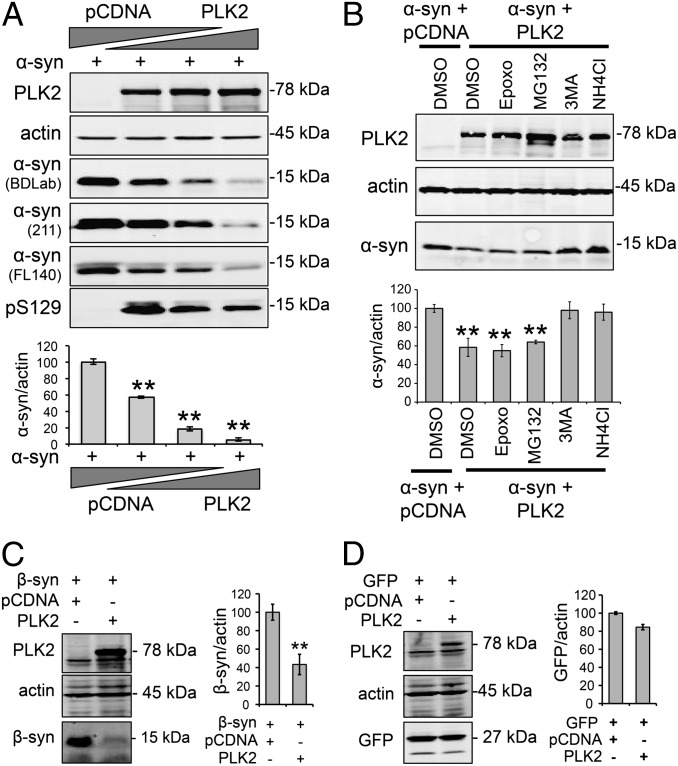
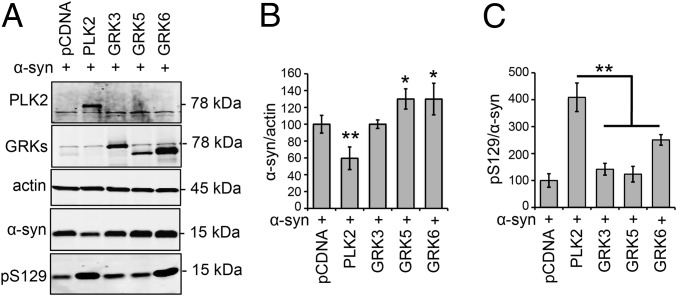
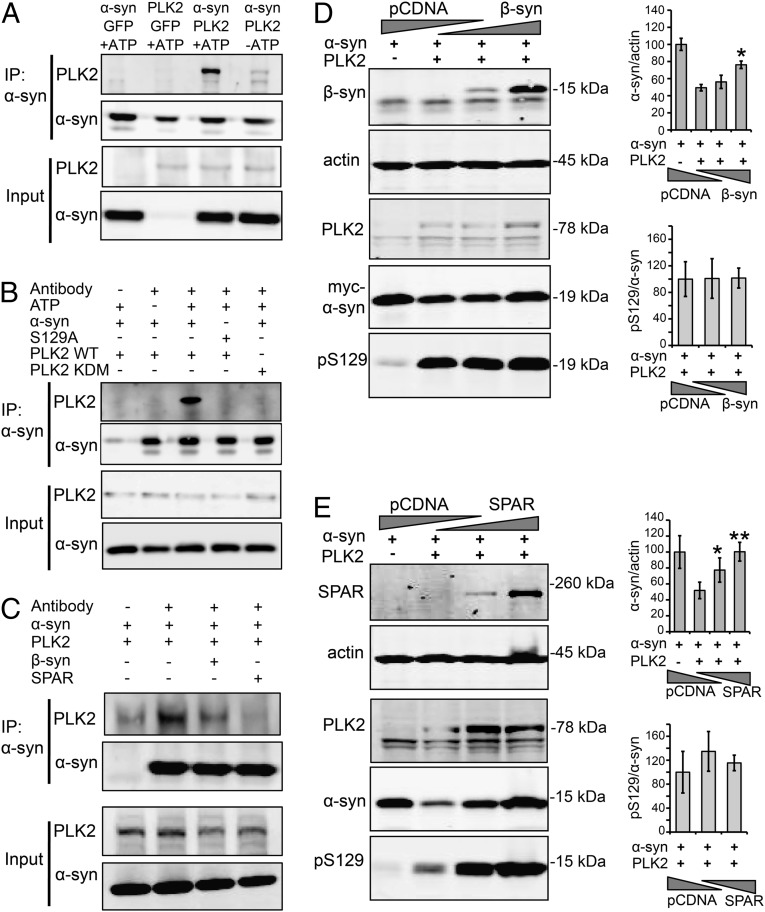
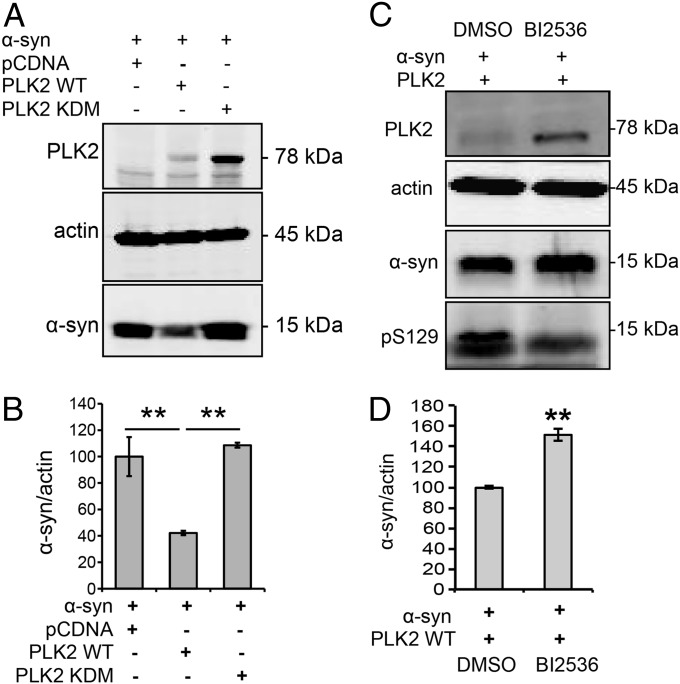
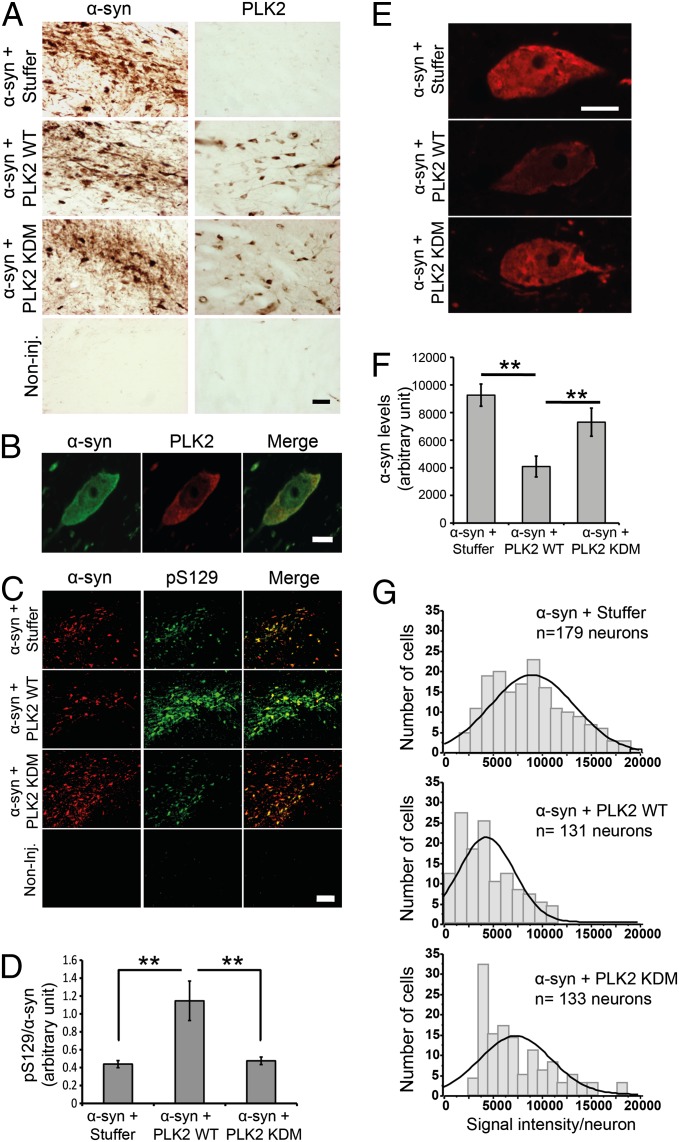
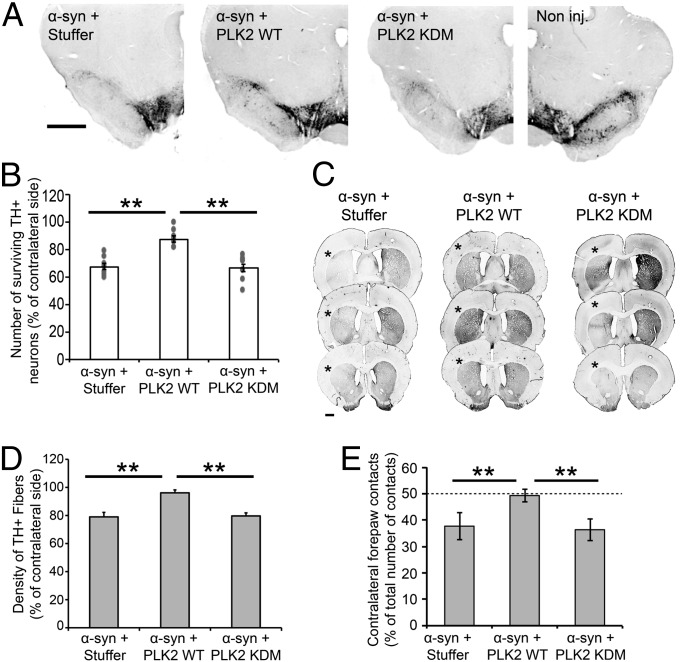
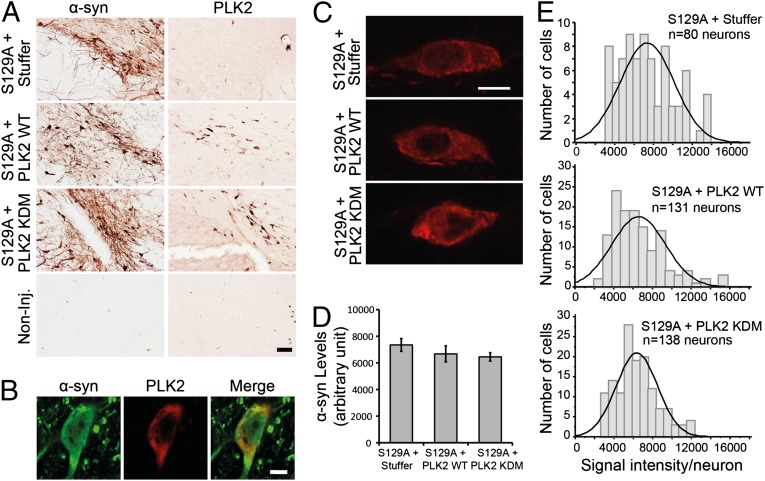
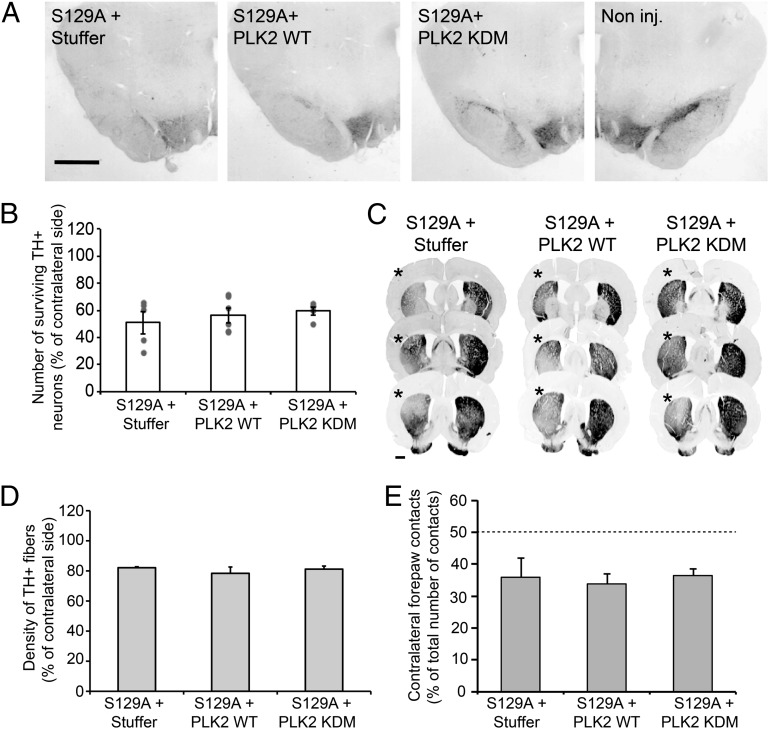
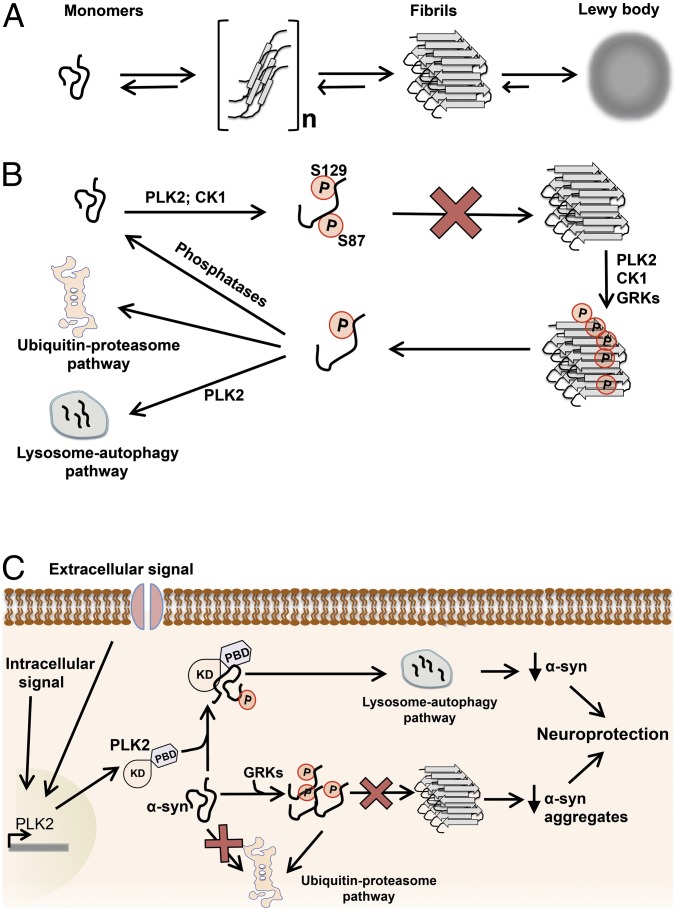
An increase in α-synuclein levels due to gene duplications/triplications or impaired degradation is sufficient to trigger its aggregation and cause familial Parkinson disease (PD). Therefore, lowering α-synuclein levels represents a viable therapeutic strategy for the treatment of PD and related synucleinopathies. Here, we report that Polo-like kinase 2 (PLK2), an enzyme up-regulated in synucleinopathy-diseased brains, interacts with, phosphorylates and enhances α-synuclein autophagic degradation in a kinase activity-dependent manner. PLK2-mediated degradation of α-synuclein requires both phosphorylation at S129 and PLK2/α-synuclein complex formation. In a rat genetic model of PD, PLK2 overexpression reduces intraneuronal human α-synuclein accumulation, suppresses dopaminergic neurodegeneration, and reverses hemiparkinsonian motor impairments induced by α-synuclein overexpression. This PLK2-mediated neuroprotective effect is also dependent on PLK2 activity and α-synuclein phosphorylation. Collectively, our findings demonstrate that PLK2 is a previously undescribed regulator of α-synuclein turnover and that modulating its kinase activity could be a viable target for the treatment of synucleinopathies.
Keywords: adeno-associated virus; animal model; serum inducible kinase.
Conflict of interest statement
Conflict of interest statement: This work has been partly supported by Merck Serono S.A., a for-profit corporation.
Figures
Comment in
-
Silencing synuclein at the synapse with PLK2.Proc Natl Acad Sci U S A. 2013 Oct 8;110(41):16293-4. doi: 10.1073/pnas.1315622110. Epub 2013 Sep 26. Proc Natl Acad Sci U S A. 2013. PMID: 24072649 Free PMC article. No abstract available.
References
-
- Lang AE, Lozano AM. Parkinson’s disease: First of two parts. N Engl J Med. 1998;339(15):1044–1053. - PubMed
-
- Lang AE, Lozano AM. Parkinson’s disease: Second of two parts. N Engl J Med. 1998;339(16):1130–1143. - PubMed
-
- Obeso JA, et al. Missing pieces in the Parkinson’s disease puzzle. Nat Med. 2010;16(6):653–661. - PubMed
-
- Lee VM, Trojanowski JQ. Mechanisms of Parkinson’s disease linked to pathological alpha-synuclein: New targets for drug discovery. Neuron. 2006;52(1):33–38. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
