

Substrate-triggered addition of dioxygen to the diferrous cofactor of aldehyde-deformylating oxygenase to form a diferric-peroxide intermediate
- PMID: 23987523
- PMCID: PMC3869994
- DOI: 10.1021/ja405047b
Substrate-triggered addition of dioxygen to the diferrous cofactor of aldehyde-deformylating oxygenase to form a diferric-peroxide intermediate
Abstract
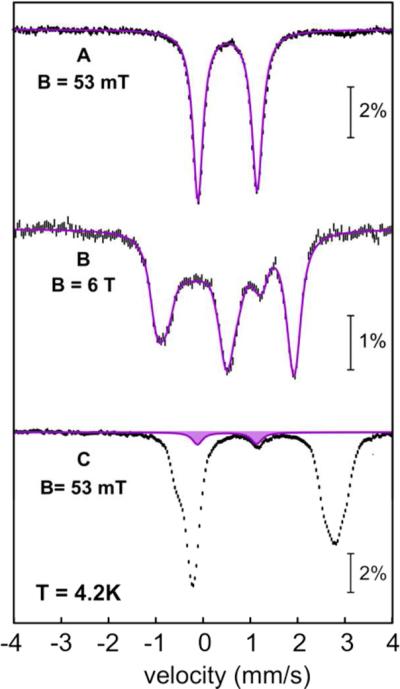
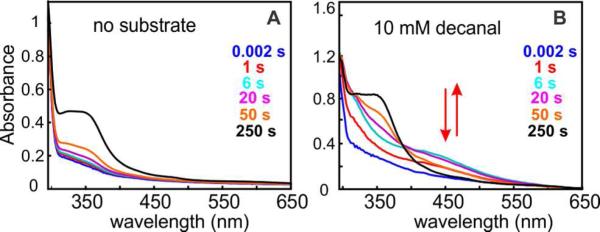
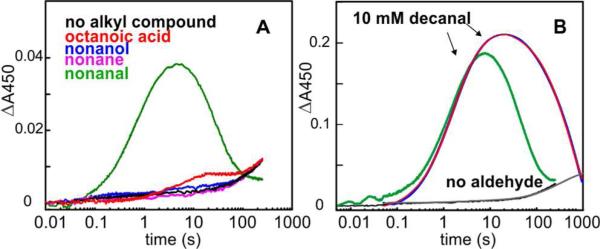
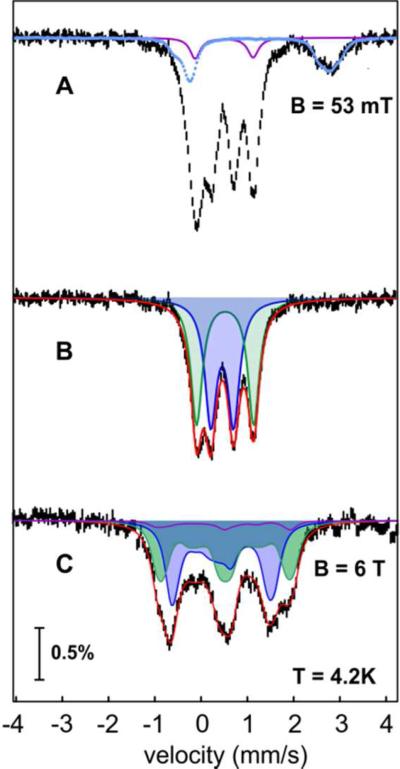
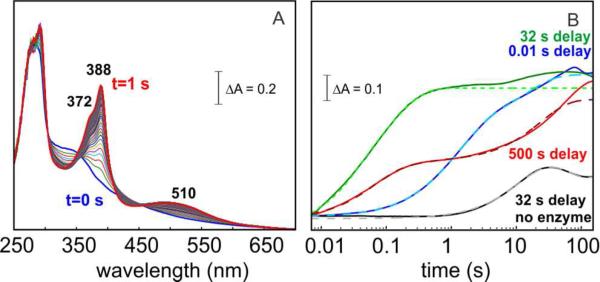
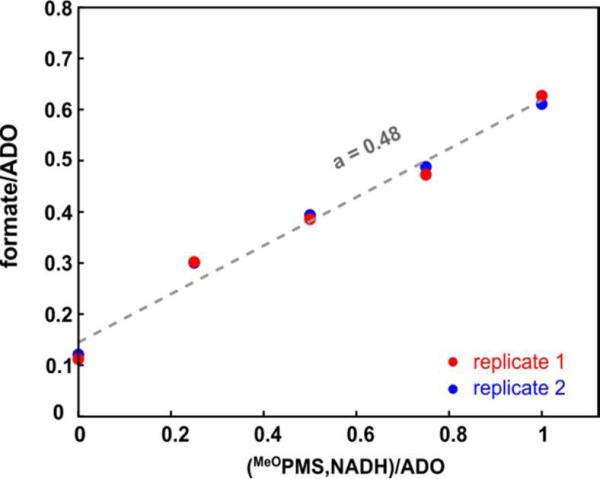
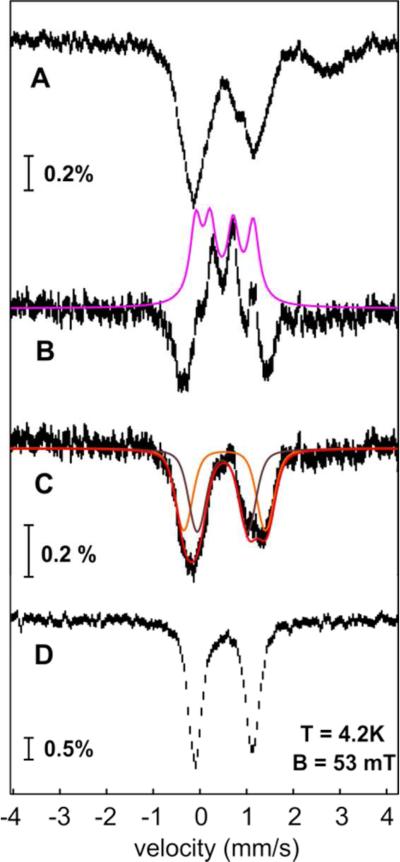
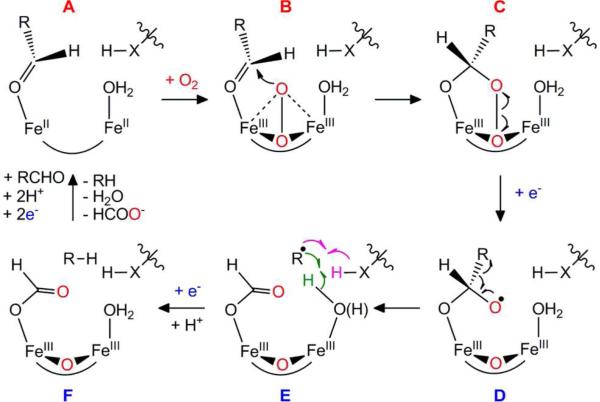
Cyanobacterial aldehyde-deformylating oxygenases (ADOs) belong to the ferritin-like diiron-carboxylate superfamily of dioxygen-activating proteins. They catalyze conversion of saturated or monounsaturated C(n) fatty aldehydes to formate and the corresponding C(n-1) alkanes or alkenes, respectively. This unusual, apparently redox-neutral transformation actually requires four electrons per turnover to reduce the O2 cosubstrate to the oxidation state of water and incorporates one O-atom from O2 into the formate coproduct. We show here that the complex of the diiron(II/II) form of ADO from Nostoc punctiforme (Np) with an aldehyde substrate reacts with O2 to form a colored intermediate with spectroscopic properties suggestive of a Fe2(III/III) complex with a bound peroxide. Its Mössbauer spectra reveal that the intermediate possesses an antiferromagnetically (AF) coupled Fe2(III/III) center with resolved subsites. The intermediate is long-lived in the absence of a reducing system, decaying slowly (t(1/2) ~ 400 s at 5 °C) to produce a very modest yield of formate (<0.15 enzyme equivalents), but reacts rapidly with the fully reduced form of 1-methoxy-5-methylphenazinium methylsulfate ((MeO)PMS) to yield product, albeit at only ~50% of the maximum theoretical yield (owing to competition from one or more unproductive pathway). The results represent the most definitive evidence to date that ADO can use a diiron cofactor (rather than a homo- or heterodinuclear cluster involving another transition metal) and provide support for a mechanism involving attack on the carbonyl of the bound substrate by the reduced O2 moiety to form a Fe2(III/III)-peroxyhemiacetal complex, which undergoes reductive O-O-bond cleavage, leading to C1-C2 radical fragmentation and formation of the alk(a/e)ne and formate products.
Figures
References
-
- Schirmer A, Rude MA, Li XZ, Popova E, del Cardayre SB. Science. 2010;329:559. - PubMed
-
- Li N, Chang WC, Warui DM, Booker SJ, Krebs C, Bollinger JM., Jr. Biochemistry. 2012;51:7908. - PubMed
-
- Reppas NB, Ridley CP. Methods and compositions for the recombinant biosynthesis of n-alkanes. Joule Unlimited Inc.; 2010. U.S. Patent 7794969.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
