

Mechanisms of altered Ca²⁺ handling in heart failure
- PMID: 23989713
- PMCID: PMC4080816
- DOI: 10.1161/CIRCRESAHA.113.301651
Mechanisms of altered Ca²⁺ handling in heart failure
Abstract
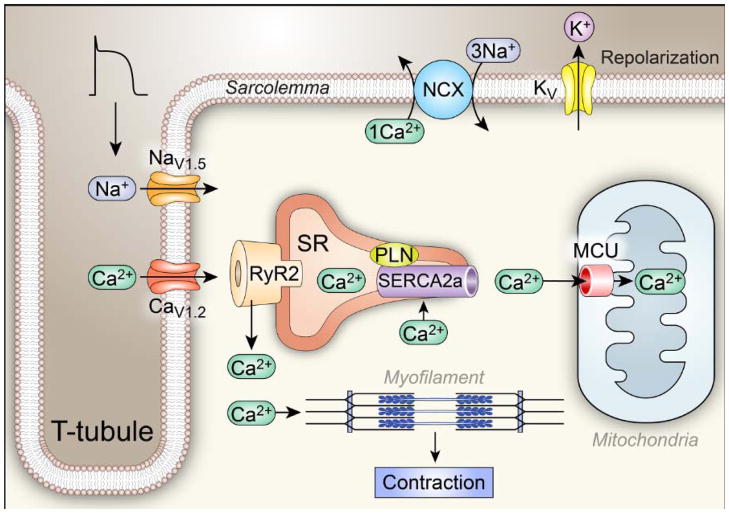
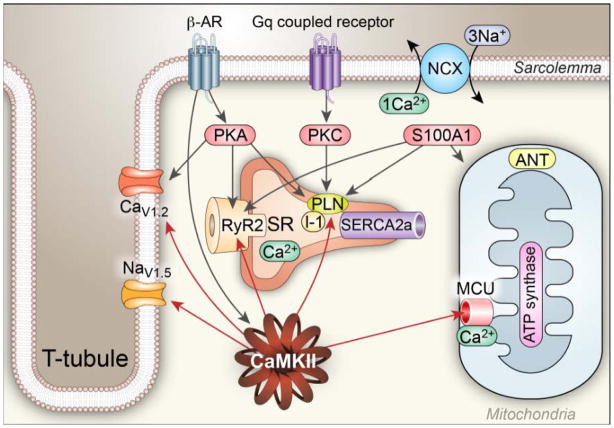
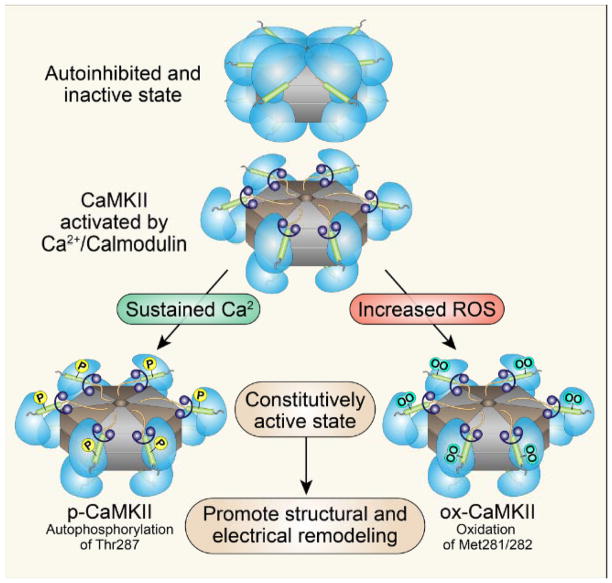
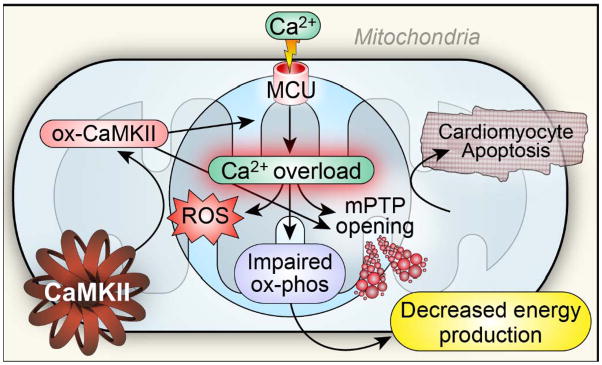
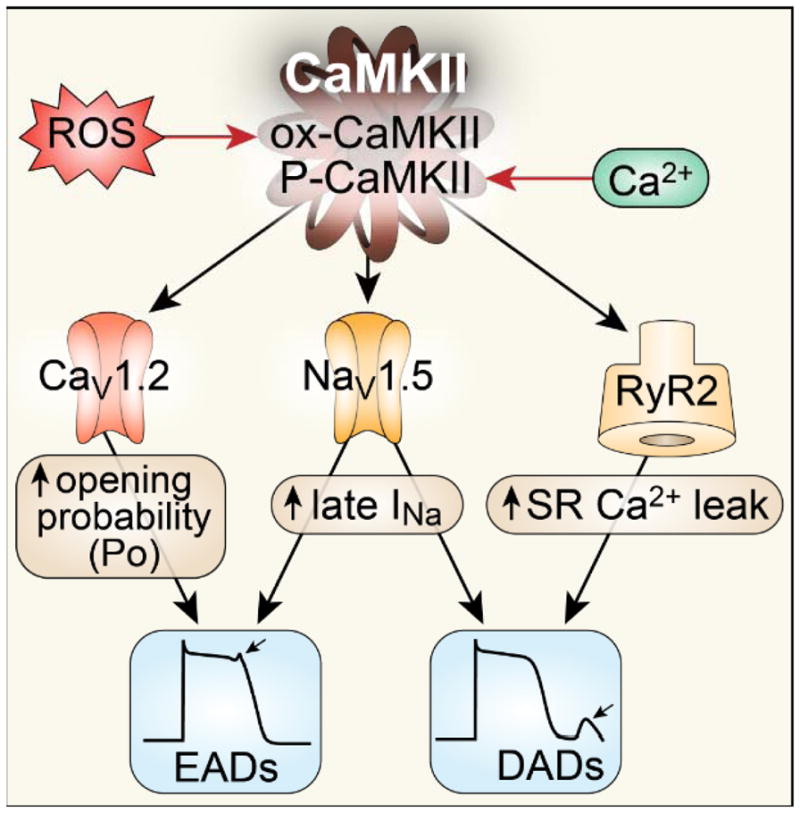
Ca²⁺ plays a crucial role in connecting membrane excitability with contraction in myocardium. The hallmark features of heart failure are mechanical dysfunction and arrhythmias; defective intracellular Ca²⁺ homeostasis is a central cause of contractile dysfunction and arrhythmias in failing myocardium. Defective Ca²⁺ homeostasis in heart failure can result from pathological alteration in the expression and activity of an increasingly understood collection of Ca²⁺ homeostatic and structural proteins, ion channels, and enzymes. This review focuses on the molecular mechanisms of defective Ca²⁺ cycling in heart failure and considers how fundamental understanding of these pathways may translate into novel and innovative therapies.
Keywords: CaMKII; calcium; excitation-contraction coupling; heart failure; mitochondria.
Figures
References
-
- Ertl G, Gaudron P, Neubauer S, Bauer B, Horn M, Hu K, Tian R. Cardiac dysfunction and development of heart failure. Eur Heart J. 1993;14 (Suppl A):33–37. - PubMed
-
- Schwinger RH, Bohm M, Muller-Ehmsen J, Uhlmann R, Schmidt U, Stablein A, Uberfuhr P, Kreuzer E, Reichart B, Eissner HJ, et al. Effect of inotropic stimulation on the negative force-frequency relationship in the failing human heart. Circulation. 1993;88:2267–2276. - PubMed
-
- Kurokawa J, Abriel H. Neurohormonal regulation of cardiac ion channels in chronic heart failure. J Cardiovasc Pharmacol. 2009;54:98–105. - PubMed
-
- Bouallegue A, Pandey NR, Srivastava AK. Camkii knockdown attenuates h2o2-induced phosphorylation of erk1/2, pkb/akt, and igf-1r in vascular smooth muscle cells. Free Radic Biol Med. 2009;47:858–866. - PubMed
-
- Oliveira PJ, Seica R, Coxito PM, Rolo AP, Palmeira CM, Santos MS, Moreno AJ. Enhanced permeability transition explains the reduced calcium uptake in cardiac mitochondria from streptozotocin-induced diabetic rats. FEBS Lett. 2003;554:511–514. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
