

DIST: direct imputation of summary statistics for unmeasured SNPs
- PMID: 23990413
- PMCID: PMC3810851
- DOI: 10.1093/bioinformatics/btt500
DIST: direct imputation of summary statistics for unmeasured SNPs
Abstract
Motivation: Genotype imputation methods are used to enhance the resolution of genome-wide association studies, and thus increase the detection rate for genetic signals. Although most studies report all univariate summary statistics, many of them limit the access to subject-level genotypes. Because such an access is required by all genotype imputation methods, it is helpful to develop methods that impute summary statistics without going through the interim step of imputing genotypes. Even when subject-level genotypes are available, due to the substantial computational cost of the typical genotype imputation, there is a need for faster imputation methods.
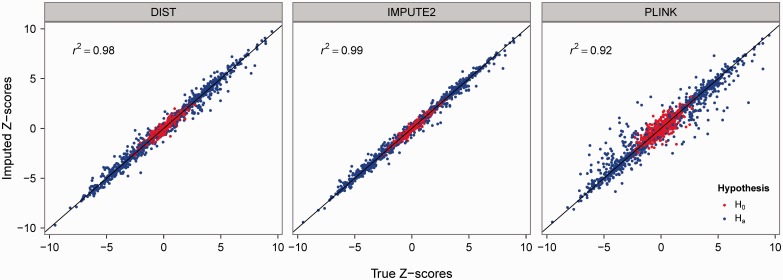
Results: Direct Imputation of summary STatistics (DIST) imputes the summary statistics of untyped variants without first imputing their subject-level genotypes. This is achieved by (i) using the conditional expectation formula for multivariate normal variates and (ii) using the correlation structure from a relevant reference population. When compared with genotype imputation methods, DIST (i) requires only a fraction of their computational resources, (ii) has comparable imputation accuracy for independent subjects and (iii) is readily applicable to the imputation of association statistics coming from large pedigree data. Thus, the proposed application is useful for a fast imputation of summary results for (i) studies of unrelated subjects, which (a) do not provide subject-level genotypes or (b) have a large size and (ii) family association studies.
Availability and implementation: Pre-compiled executables built under commonly used operating systems are publicly available at http://code.google.com/p/dist/.
Contact: dlee4@vcu.edu .
Figures
References
-
- Delaneau O, et al. A linear complexity phasing method for thousands of genomes. Nat. Methods. 2012;9:179–181. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
