

Combining quantitative genetic footprinting and trait enrichment analysis to identify fitness determinants of a bacterial pathogen
- PMID: 23990803
- PMCID: PMC3749937
- DOI: 10.1371/journal.pgen.1003716
Combining quantitative genetic footprinting and trait enrichment analysis to identify fitness determinants of a bacterial pathogen
Abstract
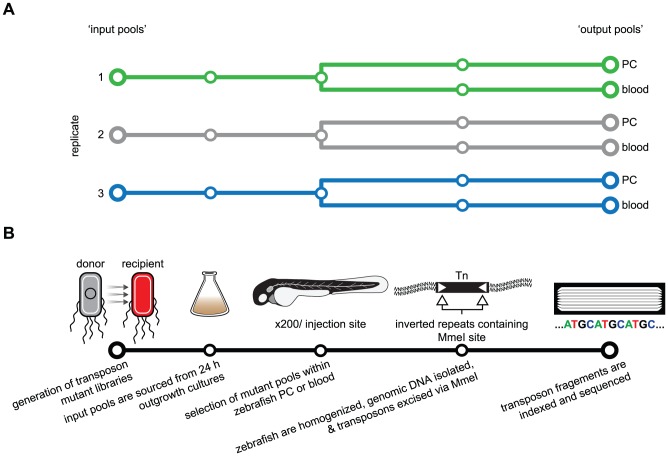
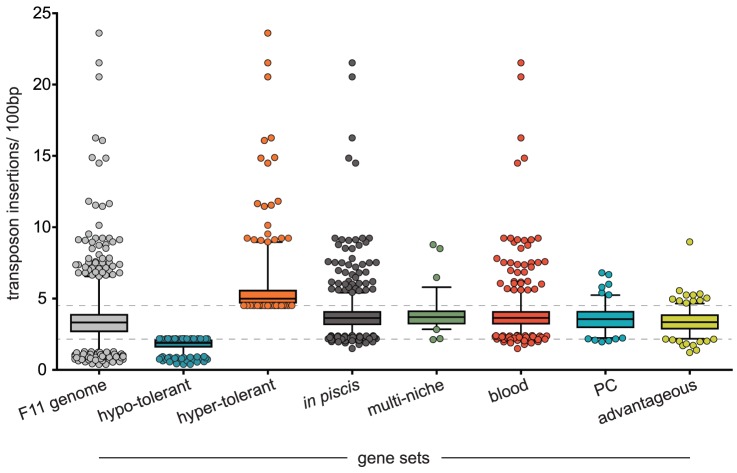
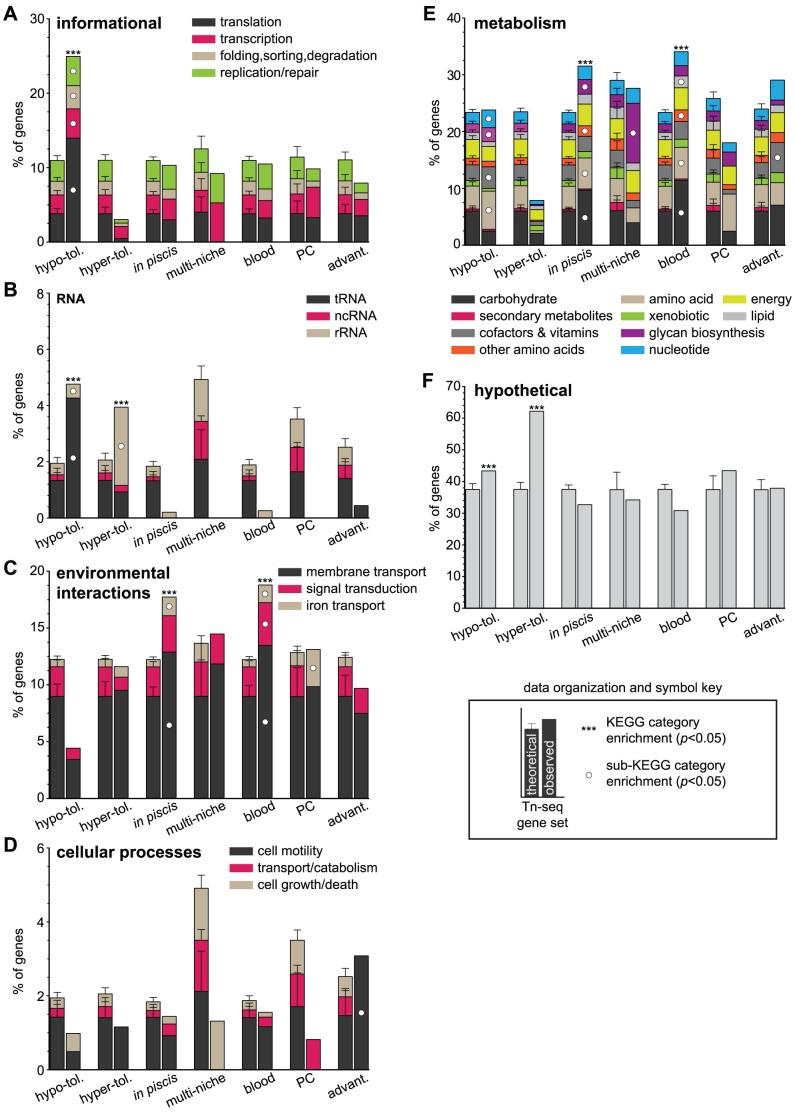
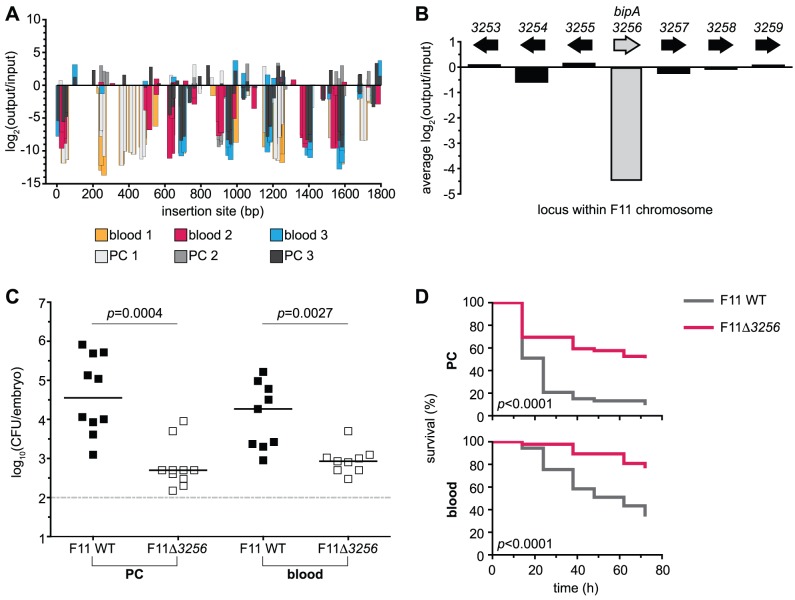
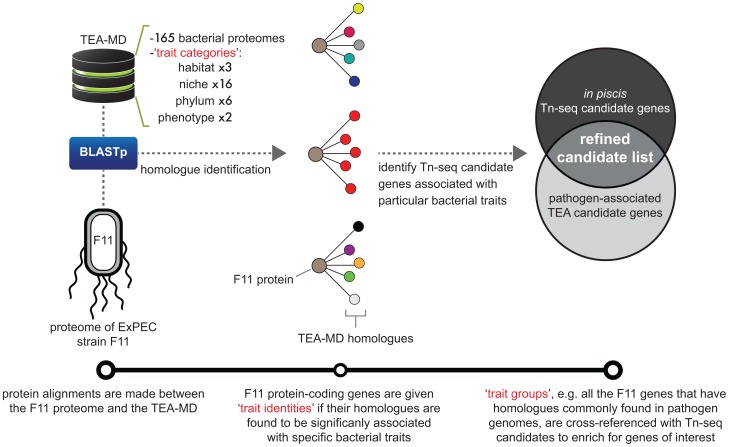
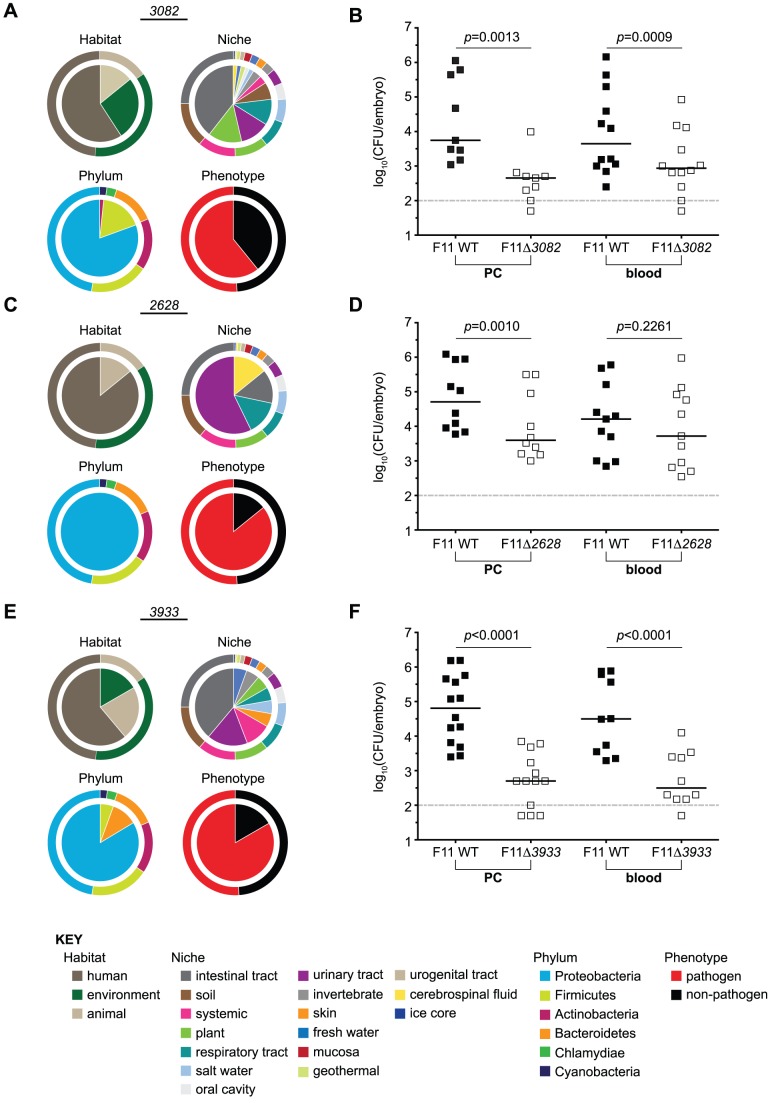
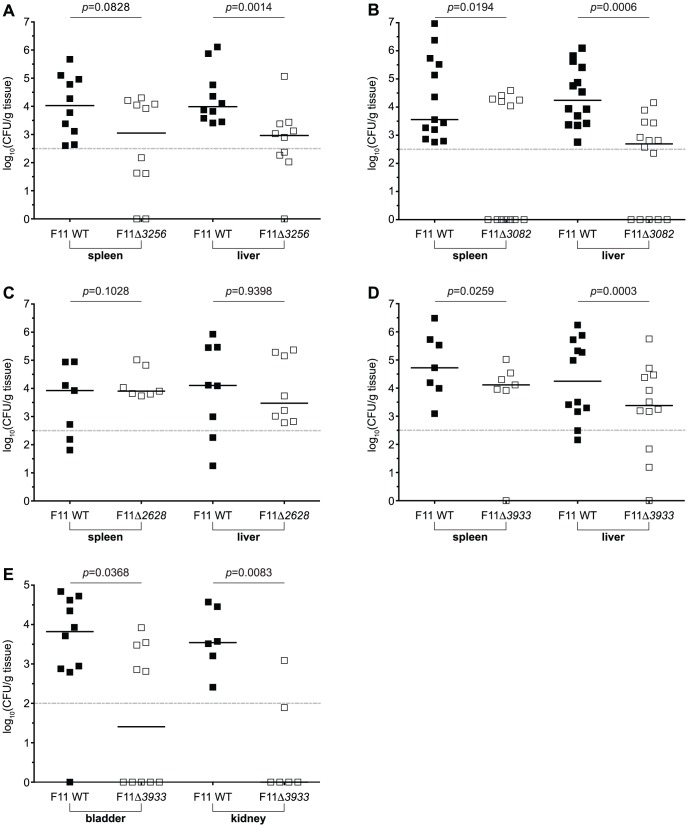
Strains of Extraintestinal Pathogenic Escherichia c oli (ExPEC) exhibit an array of virulence strategies and are a major cause of urinary tract infections, sepsis and meningitis. Efforts to understand ExPEC pathogenesis are challenged by the high degree of genetic and phenotypic variation that exists among isolates. Determining which virulence traits are widespread and which are strain-specific will greatly benefit the design of more effective therapies. Towards this goal, we utilized a quantitative genetic footprinting technique known as transposon insertion sequencing (Tn-seq) in conjunction with comparative pathogenomics to functionally dissect the genetic repertoire of a reference ExPEC isolate. Using Tn-seq and high-throughput zebrafish infection models, we tracked changes in the abundance of ExPEC variants within saturated transposon mutant libraries following selection within distinct host niches. Nine hundred and seventy bacterial genes (18% of the genome) were found to promote pathogen fitness in either a niche-dependent or independent manner. To identify genes with the highest therapeutic and diagnostic potential, a novel Trait Enrichment Analysis (TEA) algorithm was developed to ascertain the phylogenetic distribution of candidate genes. TEA revealed that a significant portion of the 970 genes identified by Tn-seq have homologues more often contained within the genomes of ExPEC and other known pathogens, which, as suggested by the first axiom of molecular Koch's postulates, is considered to be a key feature of true virulence determinants. Three of these Tn-seq-derived pathogen-associated genes--a transcriptional repressor, a putative metalloendopeptidase toxin and a hypothetical DNA binding protein--were deleted and shown to independently affect ExPEC fitness in zebrafish and mouse models of infection. Together, the approaches and observations reported herein provide a resource for future pathogenomics-based research and highlight the diversity of factors required by a single ExPEC isolate to survive within varying host environments.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Ewers C, Janssen T, Wieler LH (2003) [Avian pathogenic Escherichia coli (APEC)]. Berliner und Munchener tierarztliche Wochenschrift 116: 381–395. - PubMed
-
- Shpigel NY, Elazar S, Rosenshine I (2008) Mammary pathogenic Escherichia coli. Current opinion in microbiology 11: 60–65. - PubMed
-
- Carvallo FR, Debroy C, Baeza E, Hinckley L, Gilbert K, et al. (2010) Necrotizing pneumonia and pleuritis associated with extraintestinal pathogenic Escherichia coli in a tiger (Panthera tigris) cub. Journal of veterinary diagnostic investigation : official publication of the American Association of Veterinary Laboratory Diagnosticians, Inc 22: 136–140. - PubMed
-
- Foxman B (2010) The epidemiology of urinary tract infection. Nature reviews Urology 7: 653–660. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
