

Regulation of mitochondrial bioenergetic function by hydrogen sulfide. Part I. Biochemical and physiological mechanisms
- PMID: 23991830
- PMCID: PMC3976625
- DOI: 10.1111/bph.12369
Regulation of mitochondrial bioenergetic function by hydrogen sulfide. Part I. Biochemical and physiological mechanisms
Abstract
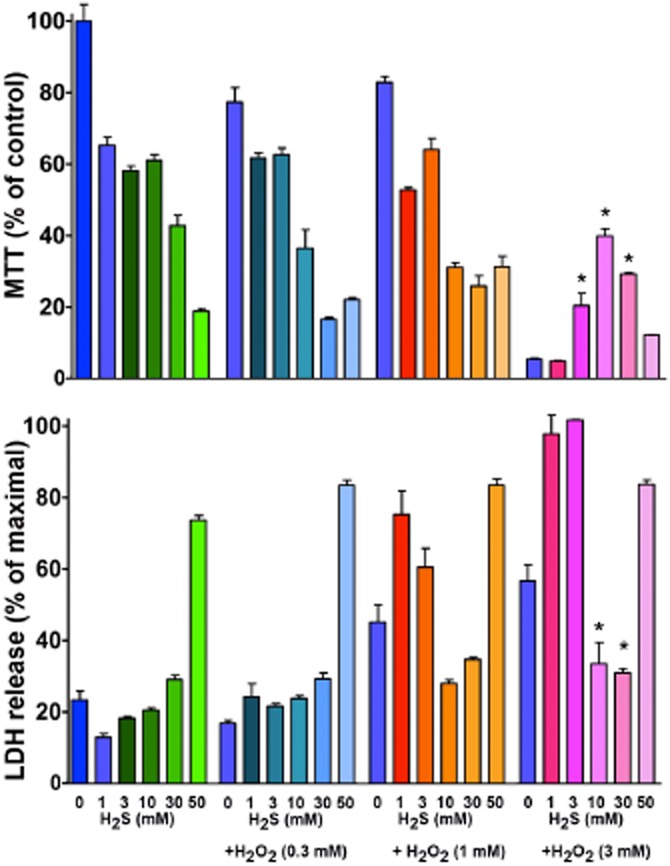
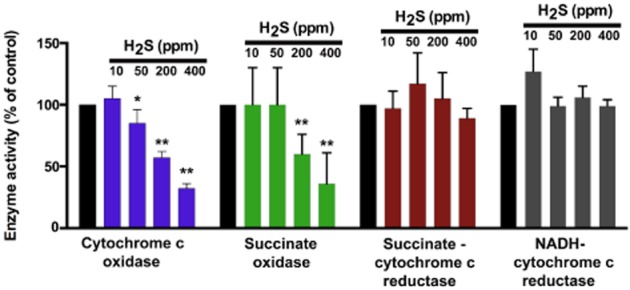
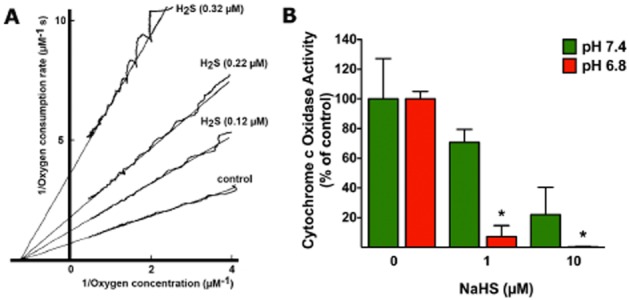
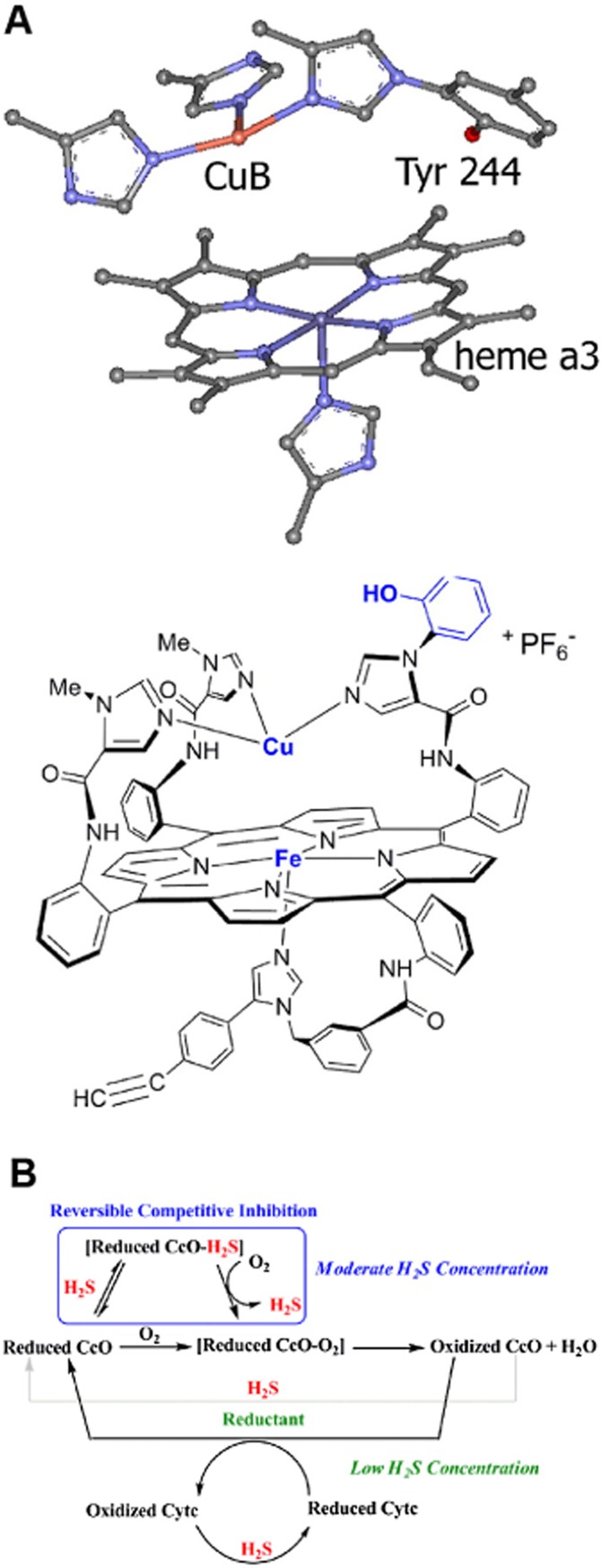
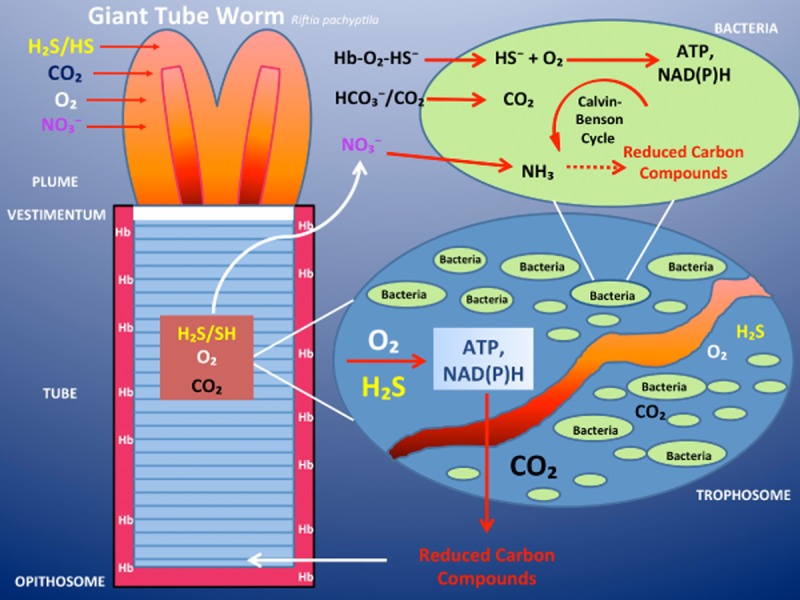
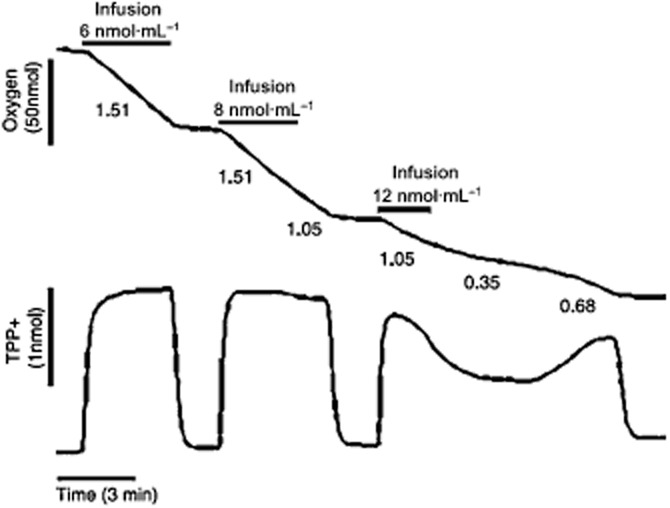
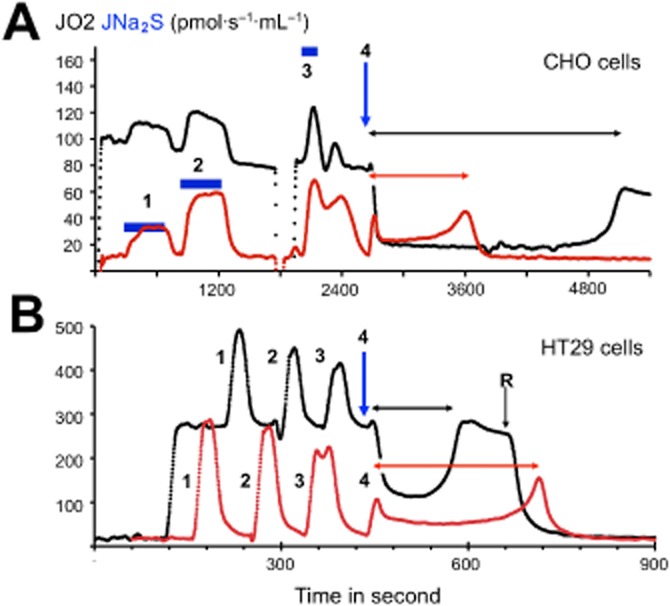
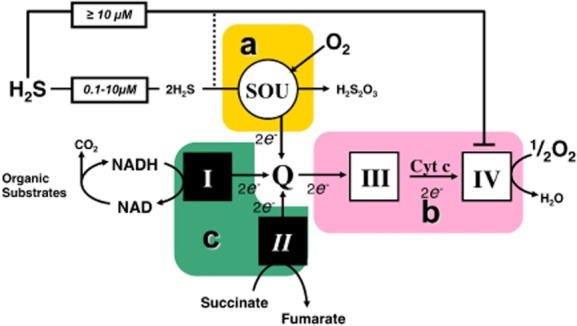
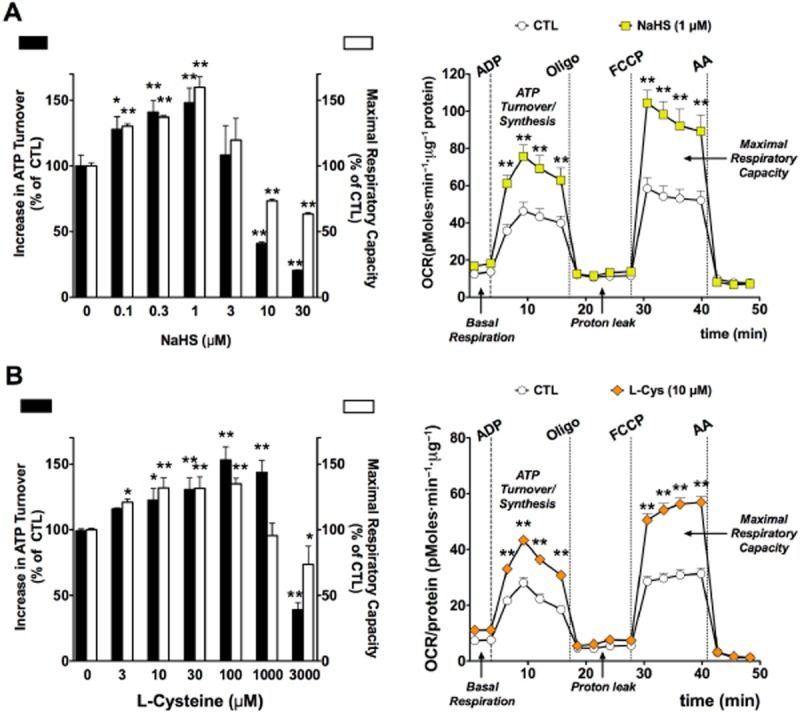
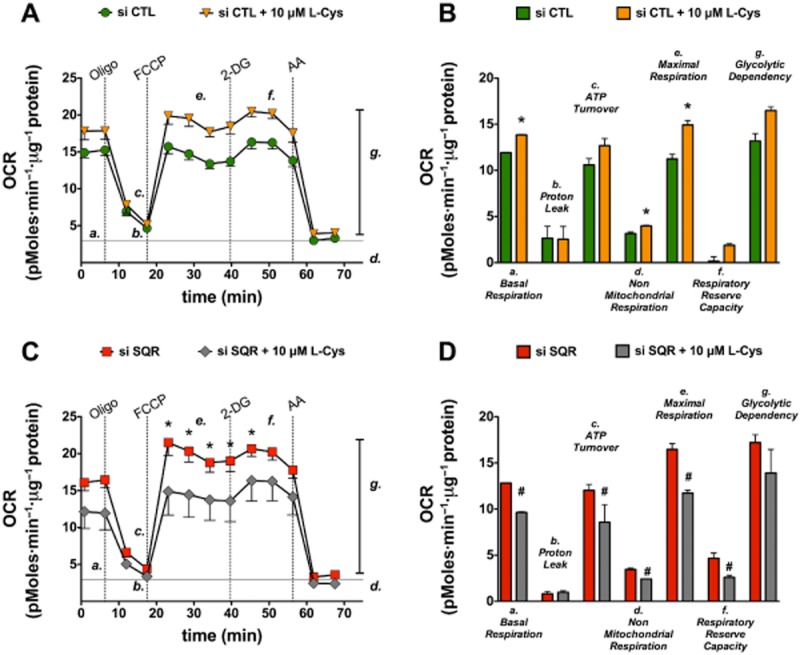
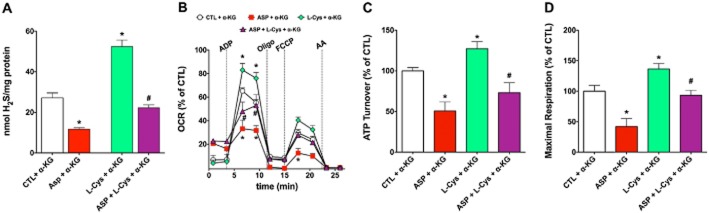
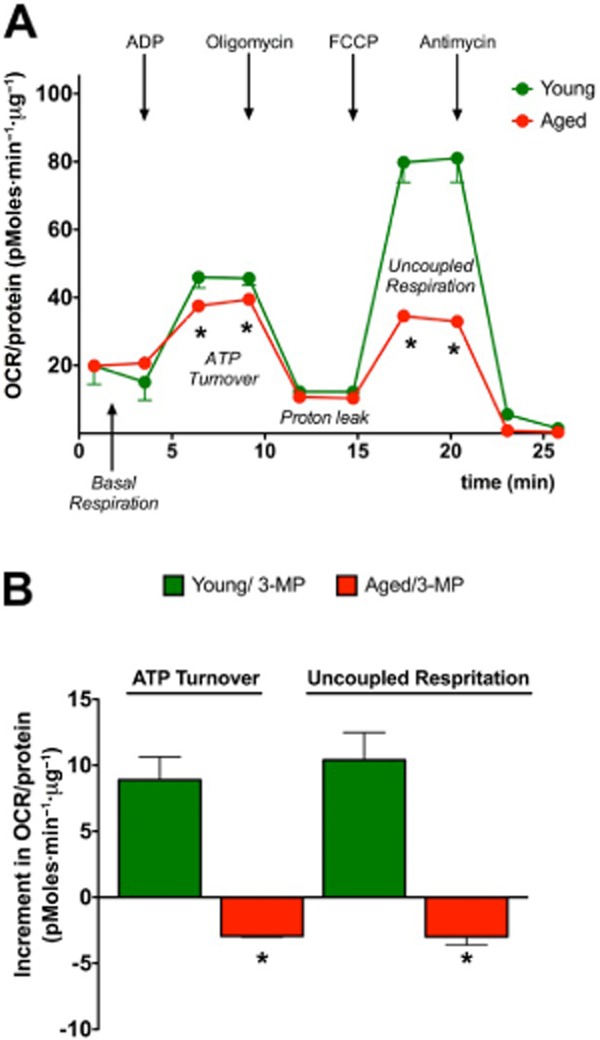
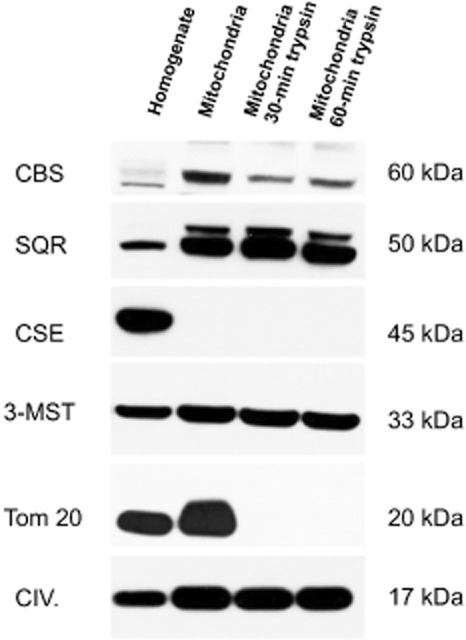
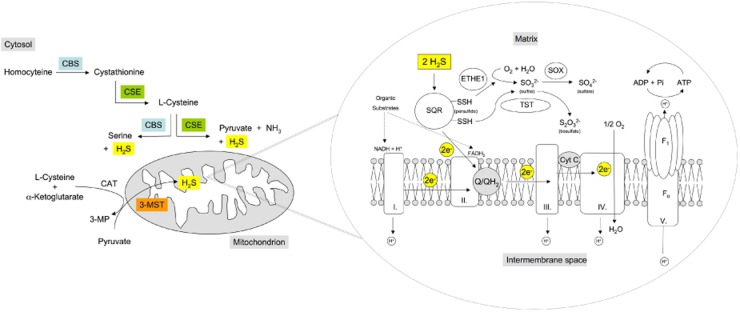
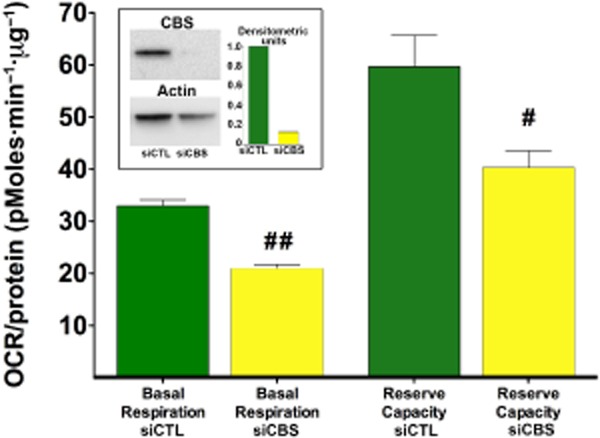
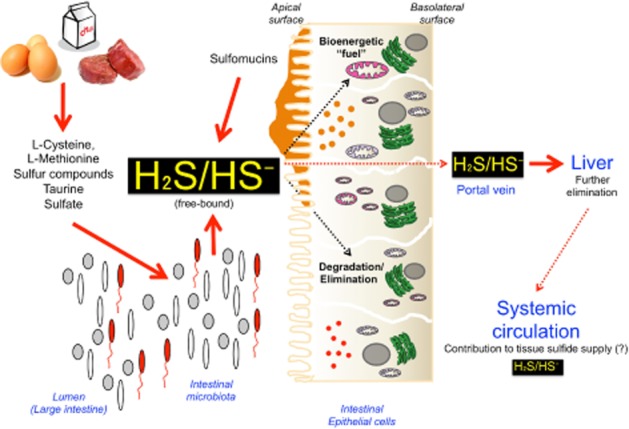
Until recently, hydrogen sulfide (H2 S) was exclusively viewed a toxic gas and an environmental hazard, with its toxicity primarily attributed to the inhibition of mitochondrial Complex IV, resulting in a shutdown of mitochondrial electron transport and cellular ATP generation. Work over the last decade established multiple biological regulatory roles of H2 S, as an endogenous gaseous transmitter. H2 S is produced by cystathionine γ-lyase (CSE), cystathionine β-synthase (CBS) and 3-mercaptopyruvate sulfurtransferase (3-MST). In striking contrast to its inhibitory effect on Complex IV, recent studies showed that at lower concentrations, H2 S serves as a stimulator of electron transport in mammalian cells, by acting as a mitochondrial electron donor. Endogenous H2 S, produced by mitochondrially localized 3-MST, supports basal, physiological cellular bioenergetic functions; the activity of this metabolic support declines with physiological aging. In specialized conditions (calcium overload in vascular smooth muscle, colon cancer cells), CSE and CBS can also associate with the mitochondria; H2 S produced by these enzymes, serves as an endogenous stimulator of cellular bioenergetics. The current article overviews the biochemical mechanisms underlying the stimulatory and inhibitory effects of H2 S on mitochondrial function and cellular bioenergetics and discusses the implication of these processes for normal cellular physiology. The relevance of H2 S biology is also discussed in the context of colonic epithelial cell physiology: colonocytes are exposed to high levels of sulfide produced by enteric bacteria, and serve as a metabolic barrier to limit their entry into the mammalian host, while, at the same time, utilizing it as a metabolic 'fuel'.
Keywords: 3-mercaptopyruvate sulfurtransferase; bioenergetics; blood vessels; cysteine; cytochrome c oxidase; free radicals; gasotransmitters; mitochondrial electron transport; nitric oxide; superoxide.
© 2013 The British Pharmacological Society.
Figures
References
-
- Attene-Ramos MS, Nava GM, Muellner MG, Wagner ED, Plewa MJ, Gaskins HR. DNA damage and toxicogenomic analyses of hydrogen sulfide in human intestinal epithelial FHs 74 Int cells. Environ Mol Mutagen. 2010;51:304–314. - PubMed
-
- Beauchamp RO, Jr, Bus JS, Popp JA, Boreiko CJ, Andjelkovich DA. A critical review of the literature on hydrogen sulfide toxicity. Crit Rev Toxicol. 1984;13:25–97. - PubMed
-
- Beltowski J. Hypoxia in the renal medulla: implications for hydrogen sulfide signaling. J Pharmacol Exp Ther. 2010;334:358–363. - PubMed
-
- Blachier F, Davila AM, Mimoun S, Benetti PH, Atanasiu C, Andriamihaja M, et al. Luminal sulfide and large intestine mucosa: friend or foe? Amino Acids. 2010;39:335–347. - PubMed
-
- Blackstone E, Morrison M, Roth MB. H2S induces a suspended animation-like state in mice. Science. 2005;308:518. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
