

The 22q11.2 deletion syndrome as a window into complex neuropsychiatric disorders over the lifespan
- PMID: 23992925
- PMCID: PMC3875621
- DOI: 10.1016/j.biopsych.2013.07.019
The 22q11.2 deletion syndrome as a window into complex neuropsychiatric disorders over the lifespan
Abstract
Evidence is rapidly accumulating that rare, recurrent copy number variants represent large effect risk factors for neuropsychiatric disorders. 22q11.2 deletion syndrome (22q11DS) (velocardiofacial syndrome or DiGeorge syndrome) is the most common known contiguous gene deletion syndrome and is associated with diverse neuropsychiatric disorders across the life span. One of the most intriguing aspects of the syndrome is the variability in clinical and cognitive presentation: children with 22q11DS have high prevalence of autism spectrum, attention deficit, and anxiety disorders, as well as psychotic-like features, and up to 30% of adolescents and adults develop schizophrenia-like psychosis. Recently, cases of early-onset Parkinson's disease in adults have been reported, collectively suggesting a role for disrupted dopaminergic neurotransmission in the observed neuropsychiatric phenotypes. There is also some evidence that 22q11DS-associated autism spectrum disorder and schizophrenia represent two unrelated phenotypic manifestations, consistent with a neuropsychiatric pleiotropy model. This genetic lesion thus provides a unique model for the discovery of specific genomic risk and (potentially) protective factors for neuropsychiatric disease. Here, we provide an overview of neuropsychiatric findings to date, which highlight the value of this syndrome in mapping the developmental trajectory of dimensional phenotypes that traverse multiple diagnostic categories. Potential sources of genetic variability that may contribute to the disorder's heterogeneous presentation are reviewed. Because of its known genetic etiology, animal models can readily be developed that recapitulate specific aspects of the syndrome. Future research directions involve translational models and potential for drug screenable targets in the context of this human model system.
Keywords: Copy number variant; dopamine; neurodevelopment; pleiotropy; schizophrenia; velocardiofacial/DiGeorge syndrome.
Copyright © 2014 Society of Biological Psychiatry. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Figures
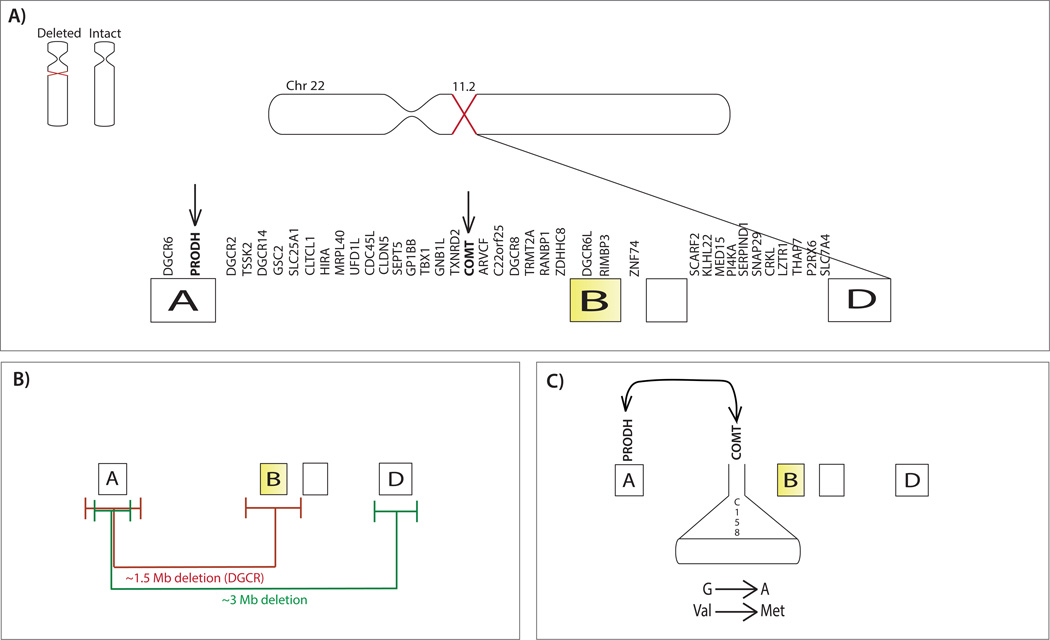
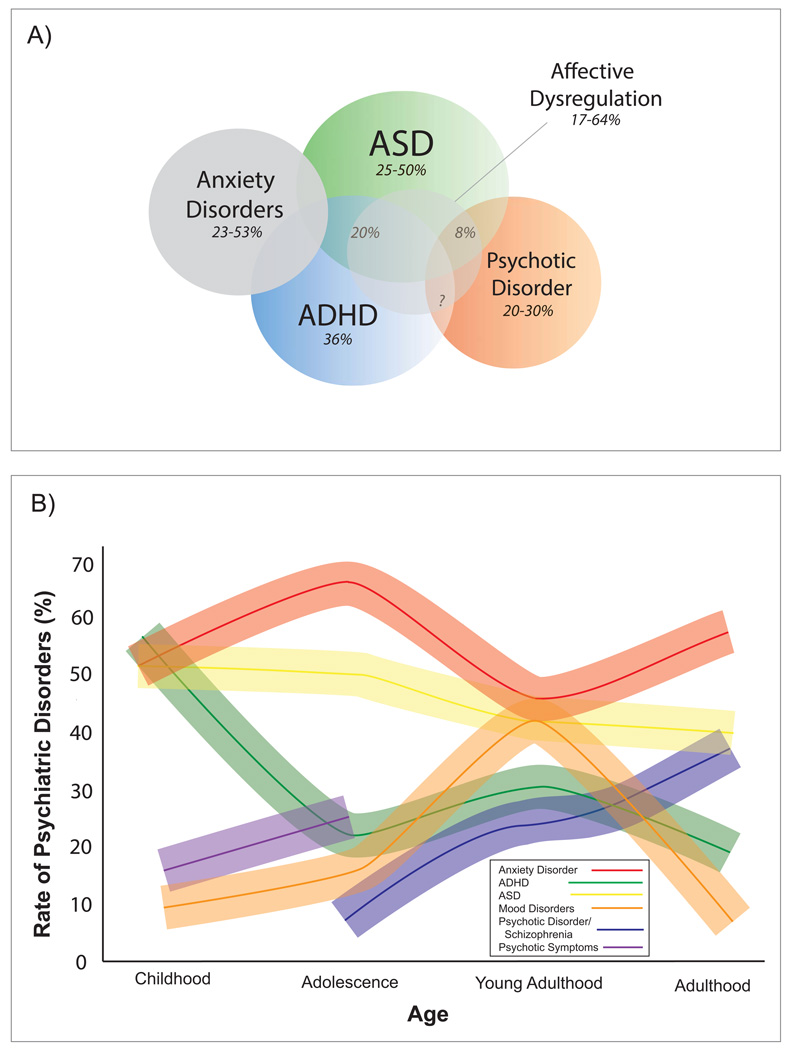
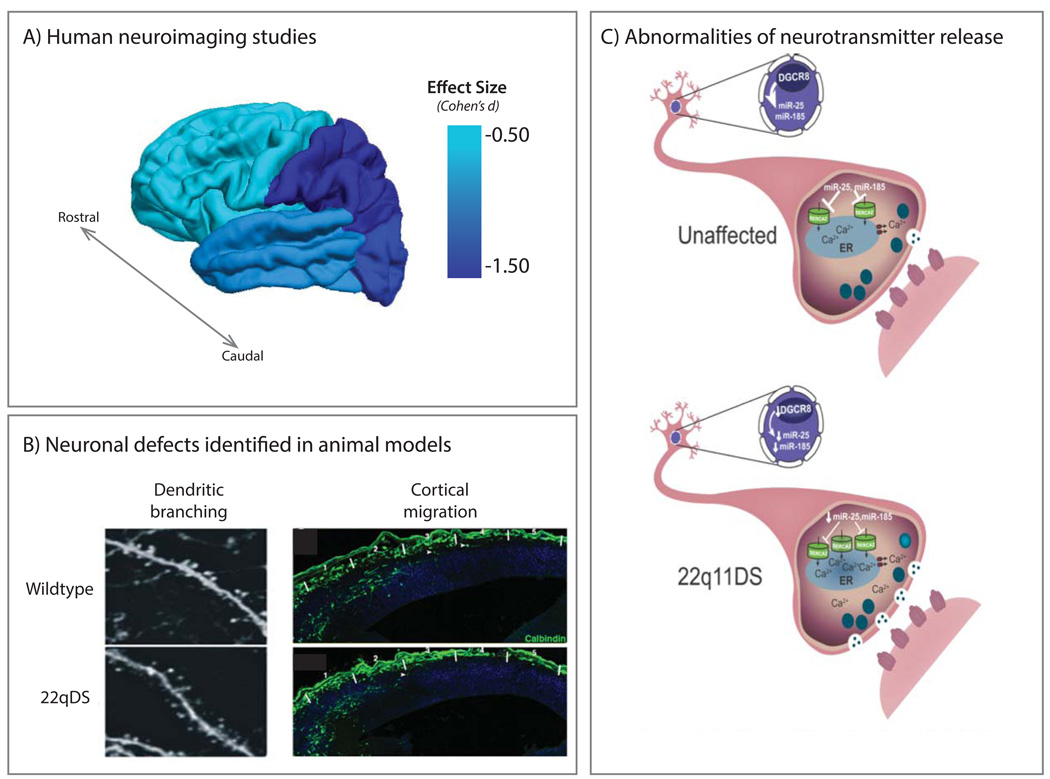
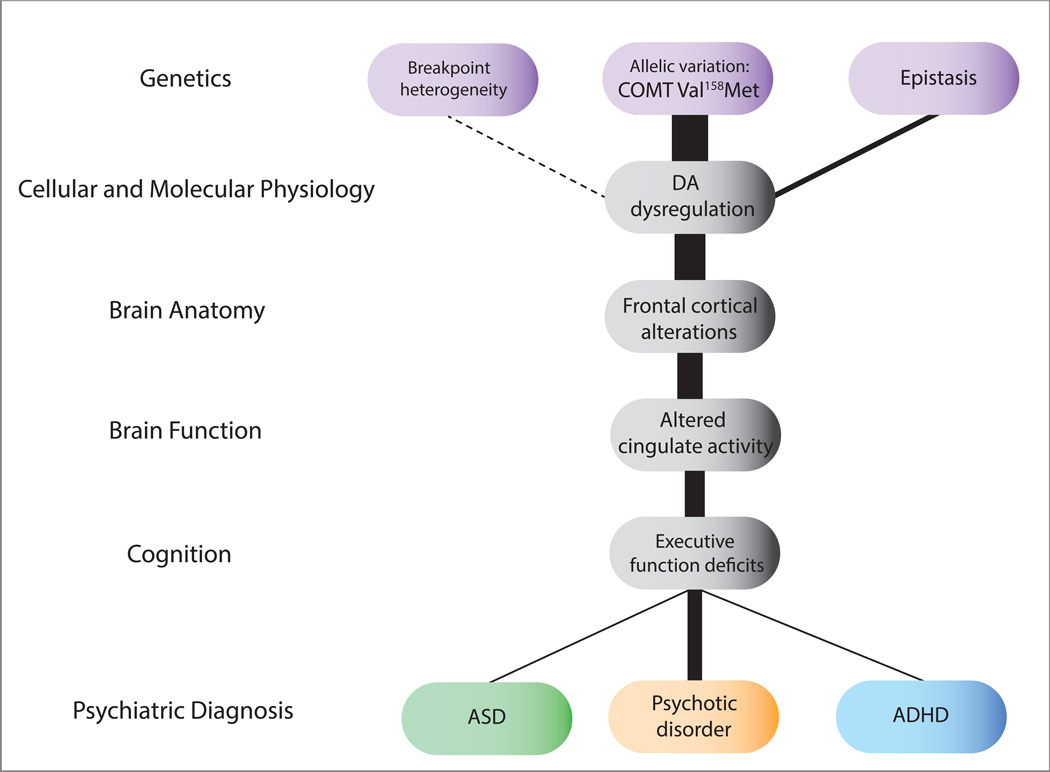
References
-
- Edelmann L, Pandita RK, Spiteri E, Funke B, Goldberg R, Palanisamy N, et al. A common molecular basis for rearrangement disorders on chromosome 22q11. Hum Mol Genet. 1999;8:1157–1167. - PubMed
-
- Murphy KC. Schizophrenia and velo-cardio-facial syndrome. Lancet. 2002;359:426–430. - PubMed
-
- Green T, Gothelf D, Glaser B, Debbané M, Frisch A, Kotler M, et al. Psychiatric Disorders and Intellectual Functioning Throughout Development in Velocardiofacial (22q11.2 Deletion) Syndrome. JAAC. 2009;48:1060–1068. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
