

Mutations in FBXL4, encoding a mitochondrial protein, cause early-onset mitochondrial encephalomyopathy
- PMID: 23993194
- PMCID: PMC3769923
- DOI: 10.1016/j.ajhg.2013.07.016
Mutations in FBXL4, encoding a mitochondrial protein, cause early-onset mitochondrial encephalomyopathy
Abstract
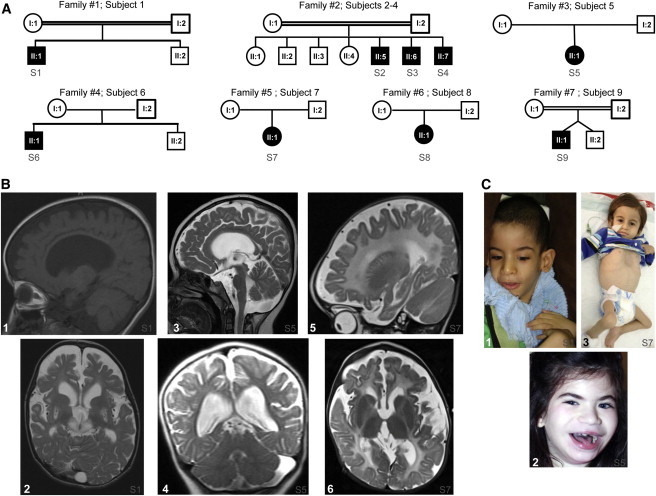
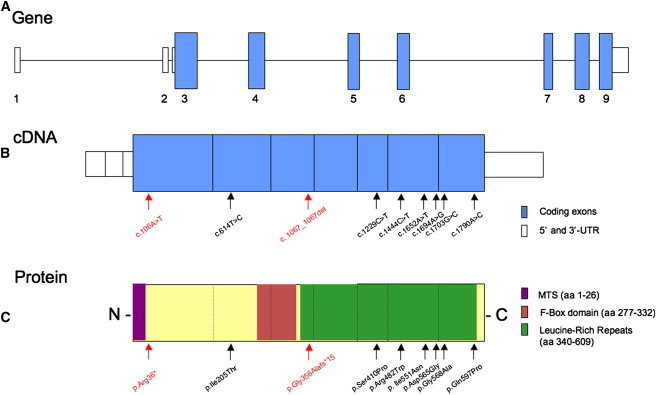
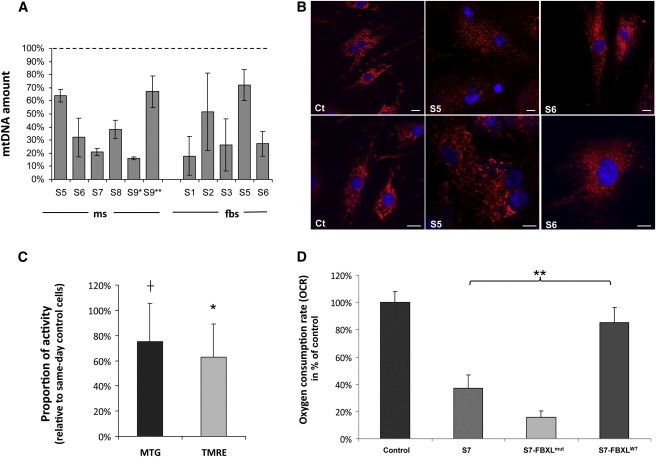
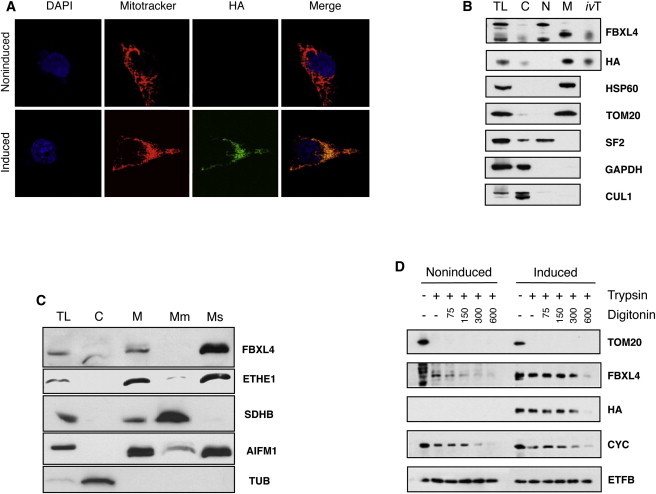
Whole-exome sequencing and autozygosity mapping studies, independently performed in subjects with defective combined mitochondrial OXPHOS-enzyme deficiencies, identified a total of nine disease-segregating FBXL4 mutations in seven unrelated mitochondrial disease families, composed of six singletons and three siblings. All subjects manifested early-onset lactic acidemia, hypotonia, and developmental delay caused by severe encephalomyopathy consistently associated with progressive cerebral atrophy and variable involvement of the white matter, deep gray nuclei, and brainstem structures. A wide range of other multisystem features were variably seen, including dysmorphism, skeletal abnormalities, poor growth, gastrointestinal dysmotility, renal tubular acidosis, seizures, and episodic metabolic failure. Mitochondrial respiratory chain deficiency was present in muscle or fibroblasts of all tested individuals, together with markedly reduced oxygen consumption rate and hyperfragmentation of the mitochondrial network in cultured cells. In muscle and fibroblasts from several subjects, substantially decreased mtDNA content was observed. FBXL4 is a member of the F-box family of proteins, some of which are involved in phosphorylation-dependent ubiquitination and/or G protein receptor coupling. We also demonstrate that FBXL4 is targeted to mitochondria and localizes in the intermembrane space, where it participates in an approximately 400 kDa protein complex. These data strongly support a role for FBXL4 in controlling bioenergetic homeostasis and mtDNA maintenance. FBXL4 mutations are a recurrent cause of mitochondrial encephalomyopathy onset in early infancy.
Copyright © 2013 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures

References
-
- Zhang W., Cui H., Wong L.J. Application of next generation sequencing to molecular diagnosis of inherited diseases. Top. Curr. Chem. 2012 Published online May 11, 2012. - PubMed
-
- Elstner M., Andreoli C., Klopstock T., Meitinger T., Prokisch H. The mitochondrial proteome database: MitoP2. Methods Enzymol. 2009;457:3–20. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- P30EY014104/EY/NEI NIH HHS/United States
- R03 DK082521/DK/NIDDK NIH HHS/United States
- R01-EY012910/EY/NEI NIH HHS/United States
- 322424/ERC_/European Research Council/International
- G20 RR030939/RR/NCRR NIH HHS/United States
- R01 EY012910/EY/NEI NIH HHS/United States
- MC_U105674181/MRC_/Medical Research Council/United Kingdom
- P30 EY014104/EY/NEI NIH HHS/United States
- R03-DK082446/DK/NIDDK NIH HHS/United States
- 1G20RR030939/RR/NCRR NIH HHS/United States
- GGP11139/TI_/Telethon/Italy
- CAPMC/ CIHR/Canada
- UL1 RR024134/RR/NCRR NIH HHS/United States
- (UL1-RR-024134/RR/NCRR NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
