

Epigenetic memory at embryonic enhancers identified in DNA methylation maps from adult mouse tissues
- PMID: 23995138
- PMCID: PMC4095776
- DOI: 10.1038/ng.2746
Epigenetic memory at embryonic enhancers identified in DNA methylation maps from adult mouse tissues
Abstract
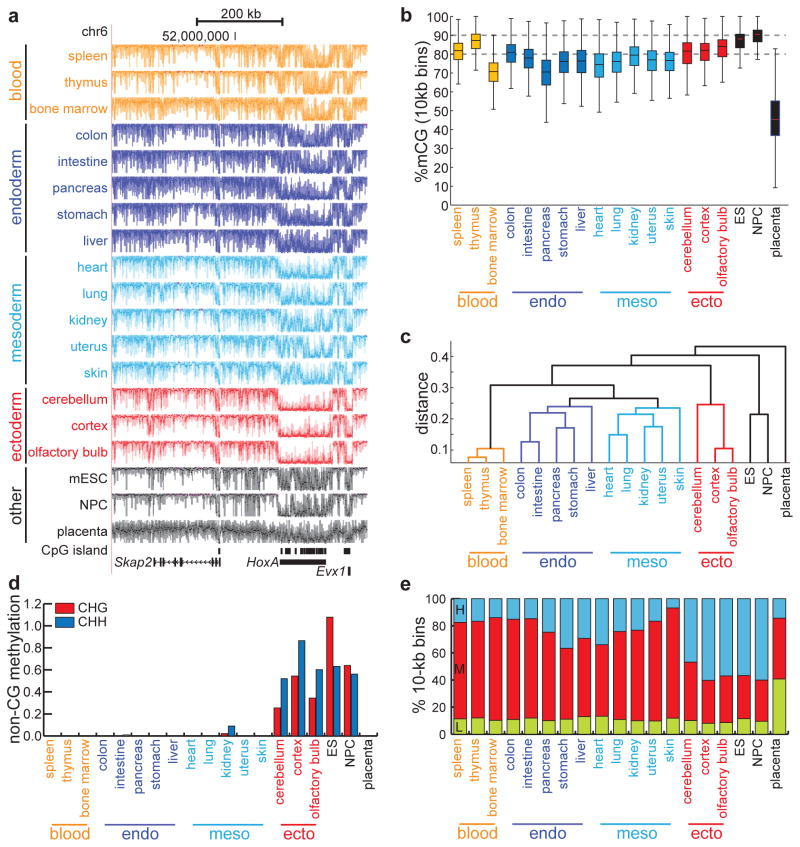
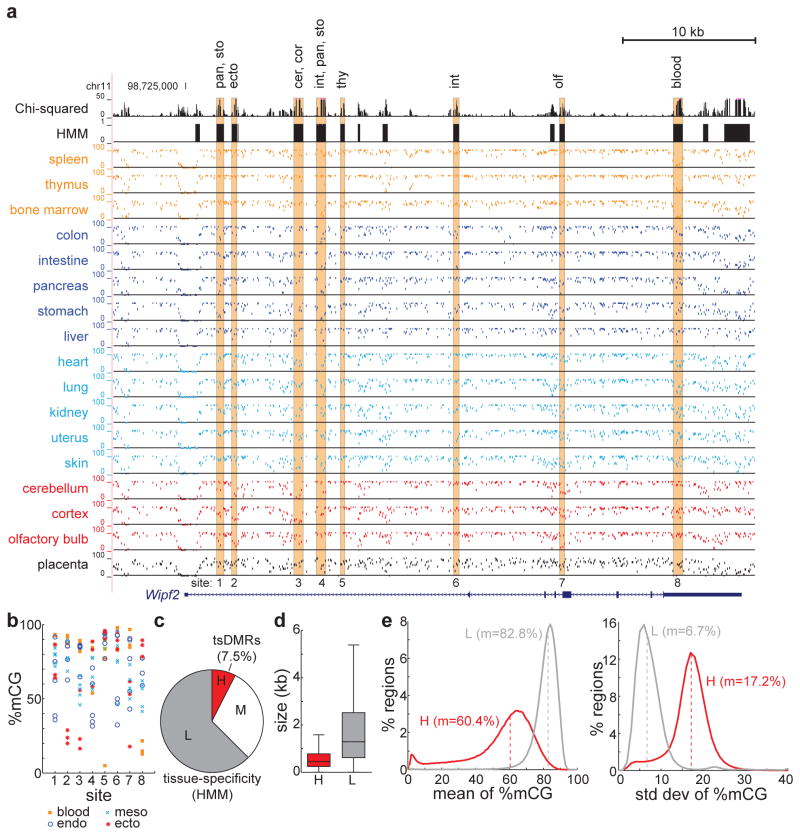
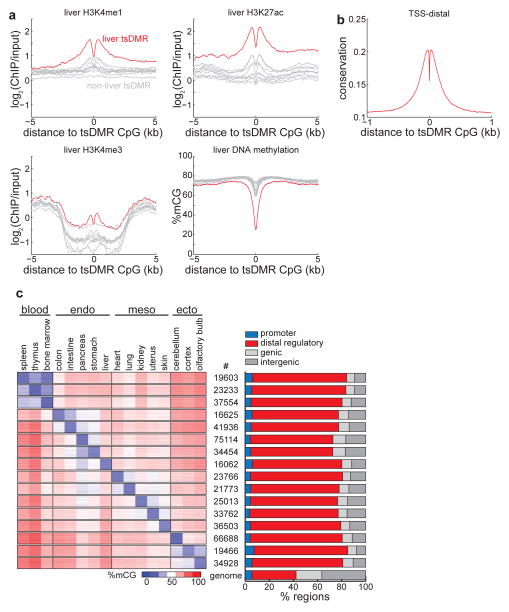
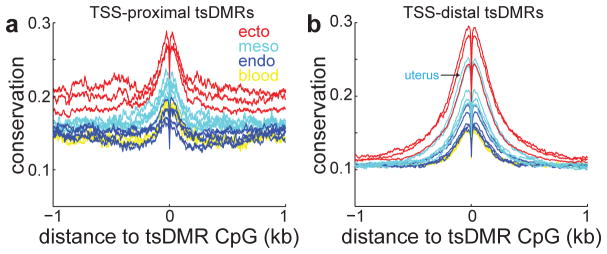
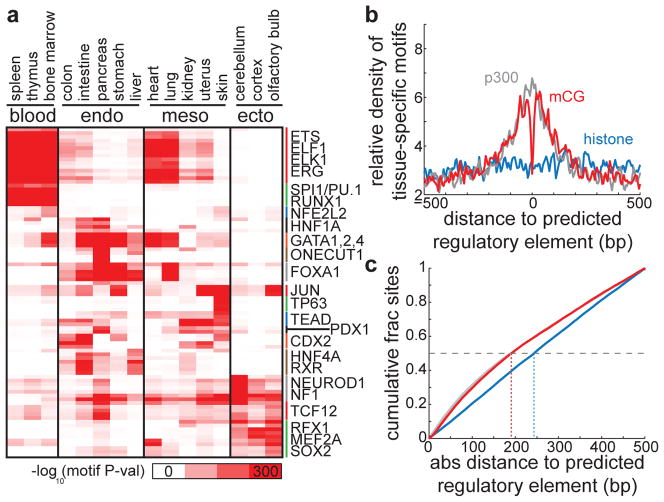
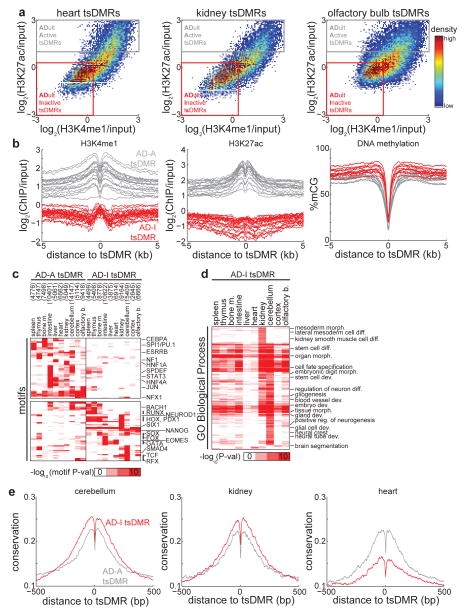
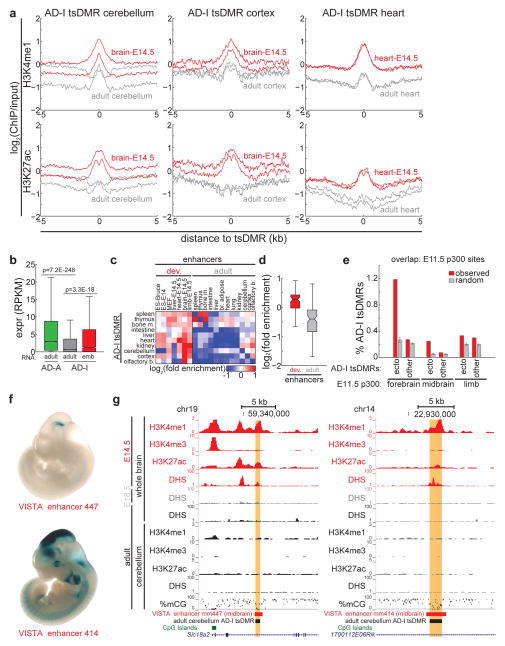
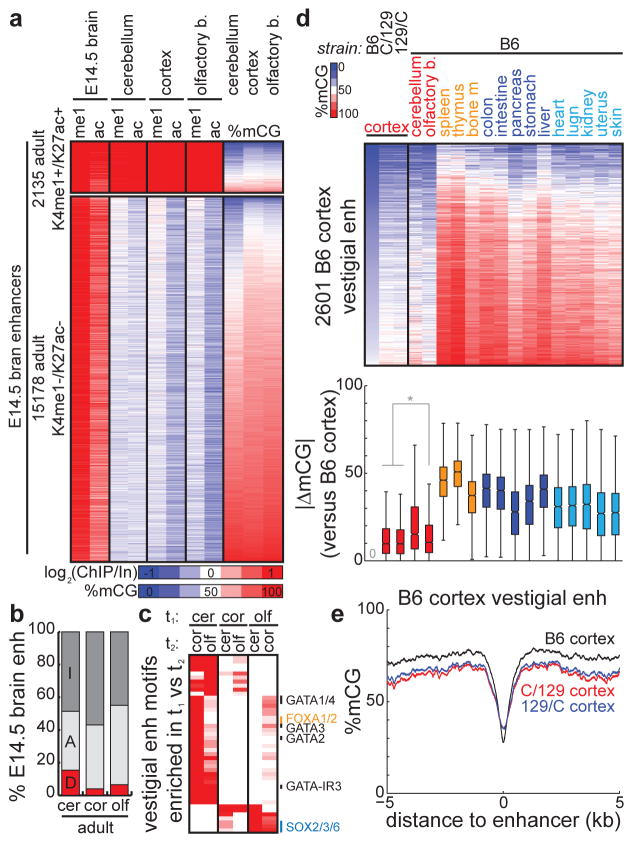
Mammalian development requires cytosine methylation, a heritable epigenetic mark of cellular memory believed to maintain a cell's unique gene expression pattern. However, it remains unclear how dynamic DNA methylation relates to cell type-specific gene expression and animal development. Here, by mapping base-resolution methylomes in 17 adult mouse tissues at shallow coverage, we identify 302,864 tissue-specific differentially methylated regions (tsDMRs) and estimate that >6.7% of the mouse genome is variably methylated. Supporting a prominent role for DNA methylation in gene regulation, most tsDMRs occur at distal cis-regulatory elements. Unexpectedly, some tsDMRs mark enhancers that are dormant in adult tissues but active in embryonic development. These 'vestigial' enhancers are hypomethylated and lack active histone modifications in adult tissues but nevertheless exhibit activity during embryonic development. Our results provide new insights into the role of DNA methylation at tissue-specific enhancers and suggest that epigenetic memory of embryonic development may be retained in adult tissues.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
-
- Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484–92. - PubMed
-
- Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. - PubMed
-
- Okano M, Li E. Genetic analyses of DNA methyltransferase genes in mouse model system. J Nutr. 2002;132:2462S–2465S. - PubMed
Publication types
MeSH terms
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
