

Human papillomavirus genome variants
- PMID: 23998342
- PMCID: PMC3979972
- DOI: 10.1016/j.virol.2013.07.018
Human papillomavirus genome variants
Abstract
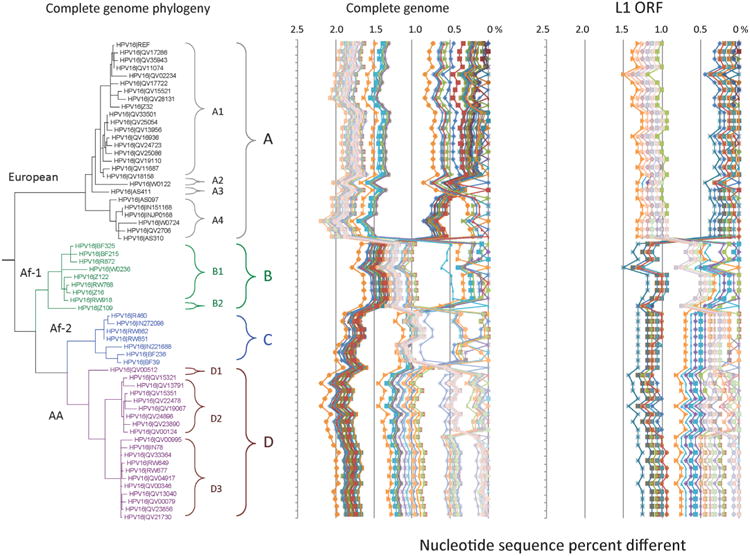
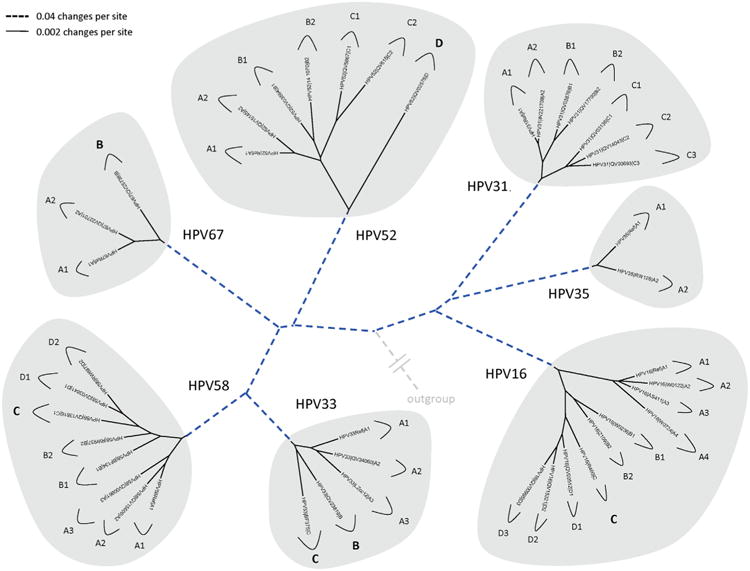
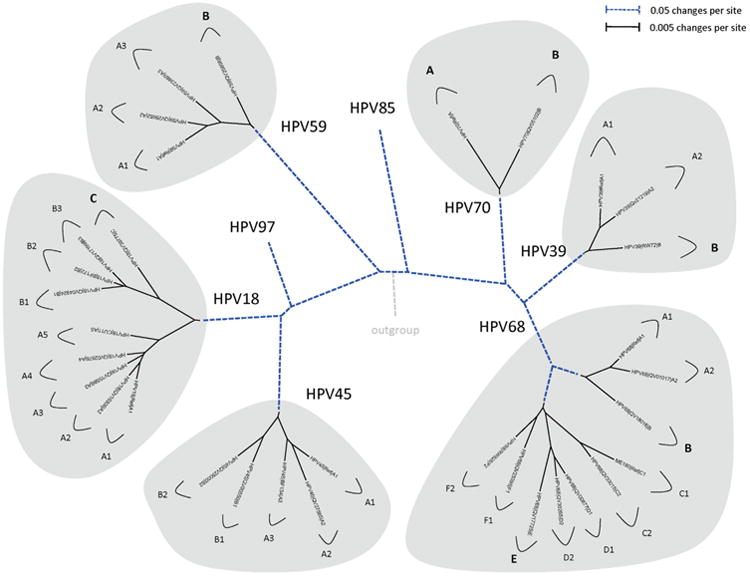
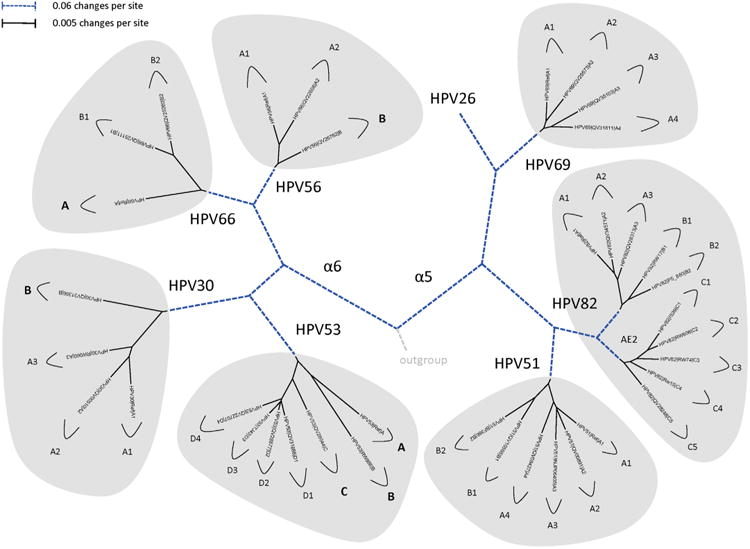
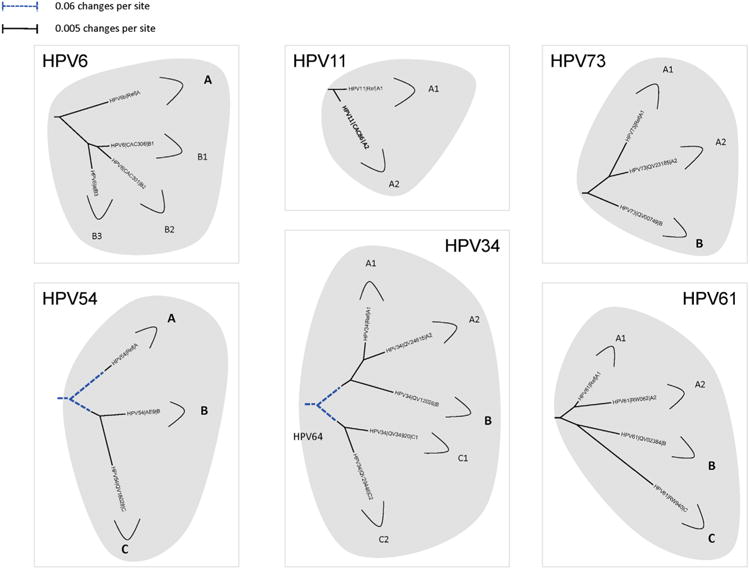
Amongst the human papillomaviruses (HPVs), the genus Alphapapillomavirus contains HPV types that are uniquely pathogenic. They can be classified into species and types based on genetic distances between viral genomes. Current circulating infectious HPVs constitute a set of viral genomes that have evolved with the rapid expansion of the human population. Viral variants were initially identified through restriction enzyme polymorphisms and more recently through sequence determination of viral fragments. Using partial sequence information, the history of variants, and the association of HPV variants with disease will be discussed with the main focus on the recent utilization of full genome sequence information for variant analyses. The use of multiple sequence alignments of complete viral genomes and phylogenetic analyses have begun to define variant lineages and sublineages using empirically defined differences of 1.0-10.0% and 0.5-1.0%, respectively. These studies provide the basis to define the genetics of HPV pathogenesis.
Keywords: Alphapapillomaviruses; HPV; HPV evolution; HPV variant lineages; Human papillomavirus variants.
© 2013 Elsevier Inc. All rights reserved.
Figures
References
-
- Alizon S, Luciani F, Regoes RR. Epidemiological and clinical consequences of within-host evolution. Trends Microbiol. 2011;19:24–32. - PubMed
-
- Arias-Pulido H, Peyton CL, Torrez-Martinez N, Anderson DN, Wheeler CM. Human papillomavirus type 18 variant lineages in United States populations characterized by sequence analysis of LCR-E6, E2, and L1 regions. Virology. 2005;338:22–34. - PubMed
-
- Barzon L, Militello V, Lavezzo E, Franchin E, Peta E, Squarzon L, Trevisan M, Pagni S, Dal Bello F, Toppo S, Palu G. Human papillomavirus genotyping by 454 next generation sequencing technology. J Clin Virol. 2011;52:93–97. - PubMed
-
- Bernard HU. Coevolution of papillomaviruses with human populations. Trends Microbiol. 1994;2:140–143. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
