

A system biology approach to identify regulatory pathways underlying the neuroendocrine control of female puberty in rats and nonhuman primates
- PMID: 23998662
- PMCID: PMC3933372
- DOI: 10.1016/j.yhbeh.2012.09.013
A system biology approach to identify regulatory pathways underlying the neuroendocrine control of female puberty in rats and nonhuman primates
Abstract
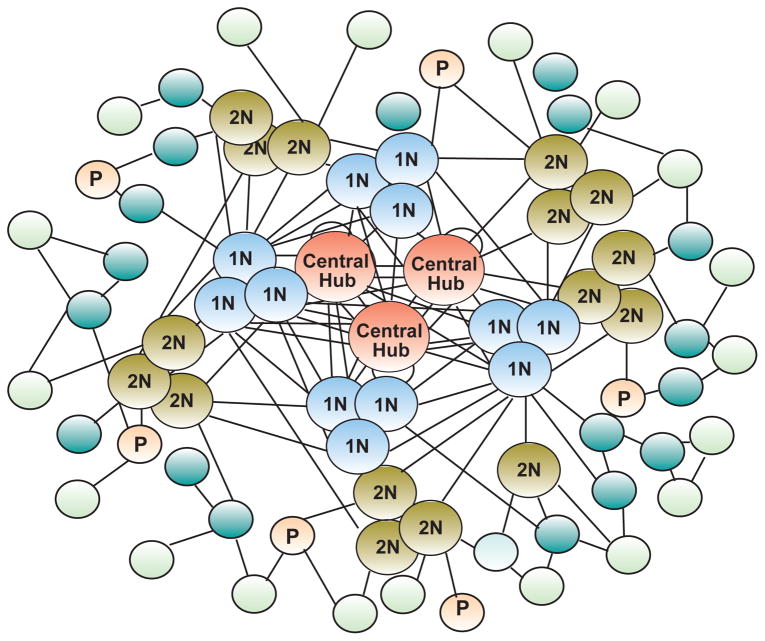
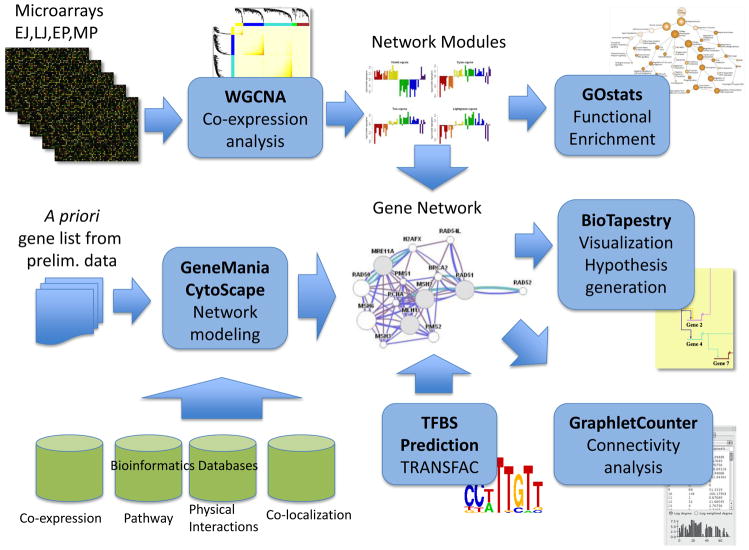
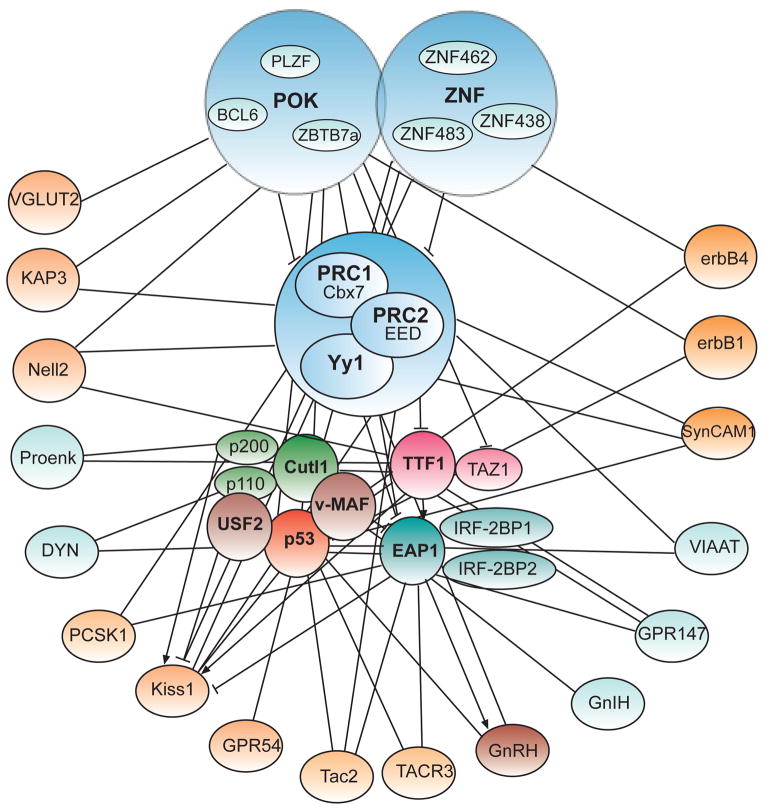
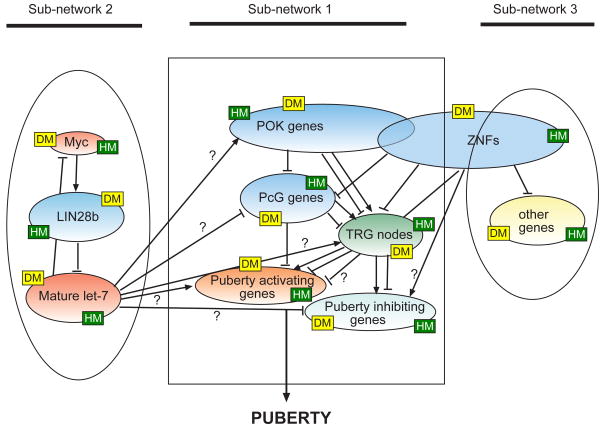
This article is part of a Special Issue "Puberty and Adolescence". Puberty is a major developmental milestone controlled by the interaction of genetic factors and environmental cues of mostly metabolic and circadian nature. An increased pulsatile release of the decapeptide gonadotropin releasing hormone (GnRH) from hypothalamic neurosecretory neurons is required for both the initiation and progression of the pubertal process. This increase is brought about by coordinated changes that occur in neuronal and glial networks associated with GnRH neurons. These changes ultimately result in increased neuronal and glial stimulatory inputs to the GnRH neuronal network and a reduction of transsynaptic inhibitory influences. While some of the major players controlling pubertal GnRH secretion have been identified using gene-centric approaches, much less is known about the system-wide control of the overall process. Because the pubertal activation of GnRH release involves a diversity of cellular phenotypes, and a myriad of intracellular and cell-to-cell signaling molecules, it appears that the overall process is controlled by a highly coordinated and interactive regulatory system involving hundreds, if not thousands, of gene products. In this article we will discuss emerging evidence suggesting that these genes are arranged as functionally connected networks organized, both internally and across sub-networks, in a hierarchical fashion. According to this concept, the core of these networks is composed of transcriptional regulators that, by directing expression of downstream subordinate genes, provide both stability and coordination to the cellular networks involved in initiating the pubertal process. The integrative response of these gene networks to external inputs is postulated to be coordinated by epigenetic mechanisms.
Keywords: Female puberty; Gene networks; Glial–neuronal communication; Hypothalamus; Neuroendocrine control; Neurotransmission; Systems biology; Transcriptional regulation.
Copyright © 2012 Elsevier Inc. All rights reserved.
Figures
References
-
- Alexa A, Rahnenfuhrer J, Lengauer T. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics. 2006;22:1600–1607. - PubMed
-
- Artenstein AW, Opal SM. Proprotein convertases in health and disease. N Engl J Med. 2011;365:2507–2518. - PubMed
-
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. - PMC - PubMed
-
- Basso K, Margolin AA, Stolovitzky G, Klein U, Dalla-Favera R, Califano A. Reverse engineering of regulatory networks in human B cells. Nat Genet. 2005;37:382–390. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
