

Tissue- and cell-type-specific manifestations of heteroplasmic mtDNA 3243A>G mutation in human induced pluripotent stem cell-derived disease model
- PMID: 24003133
- PMCID: PMC3780874
- DOI: 10.1073/pnas.1311660110
Tissue- and cell-type-specific manifestations of heteroplasmic mtDNA 3243A>G mutation in human induced pluripotent stem cell-derived disease model
Abstract
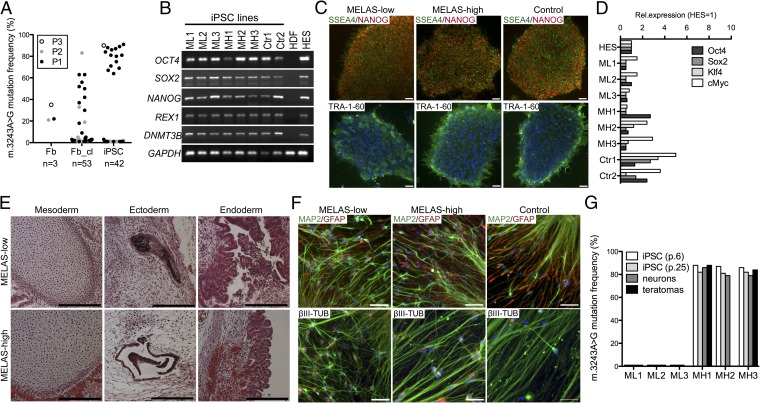
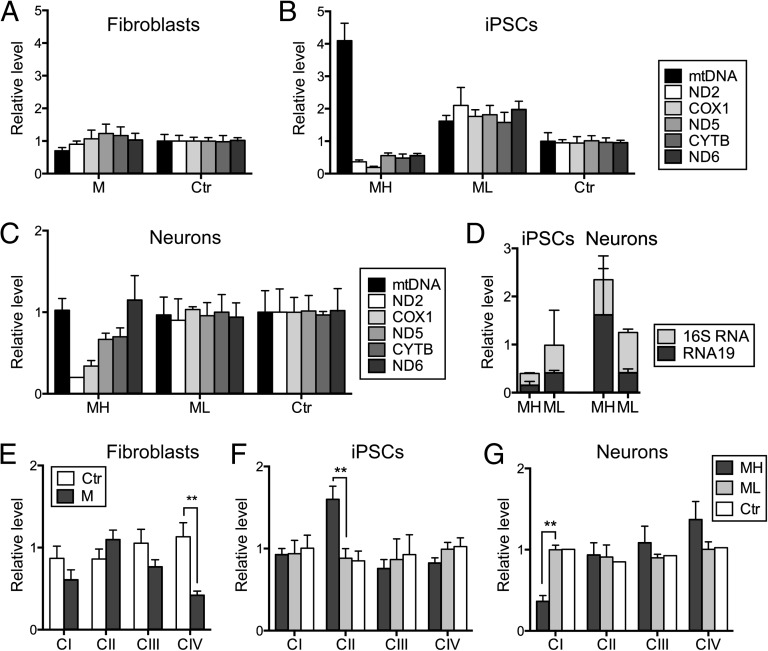
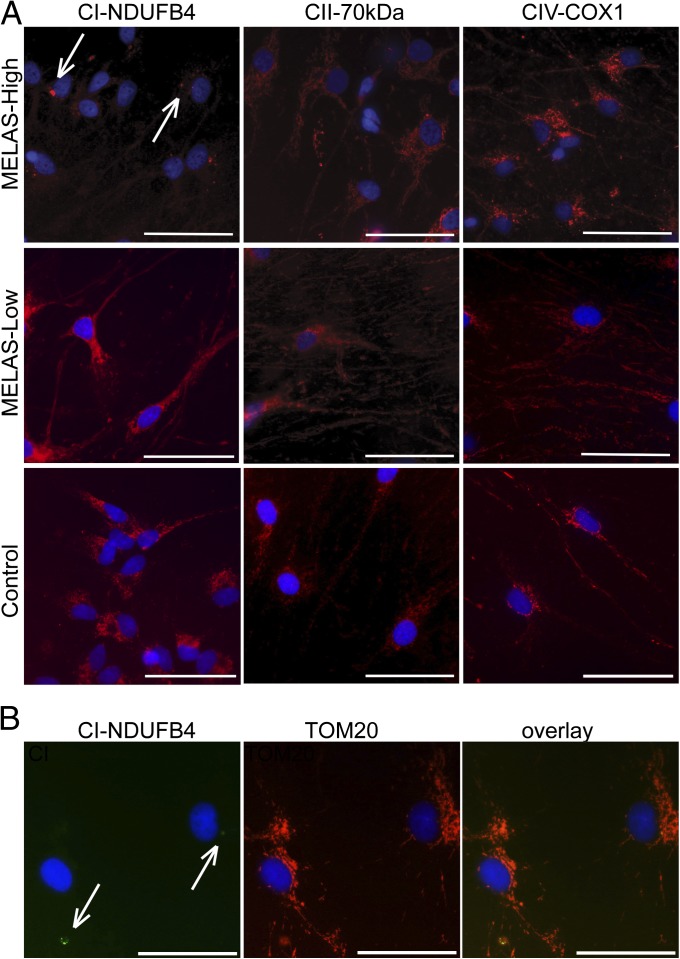
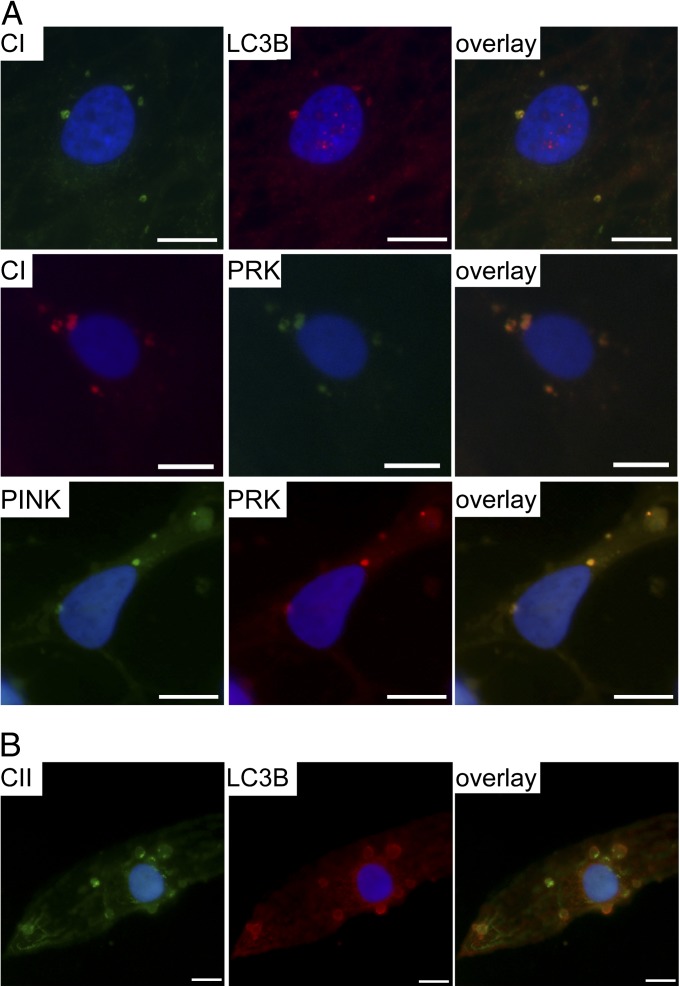
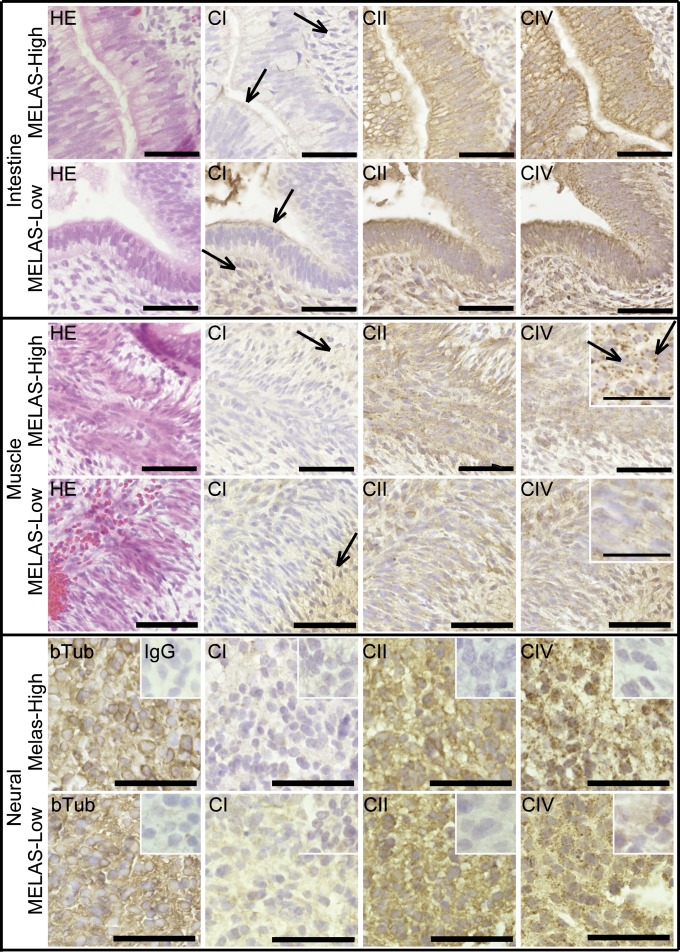
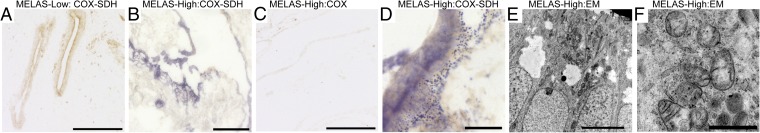
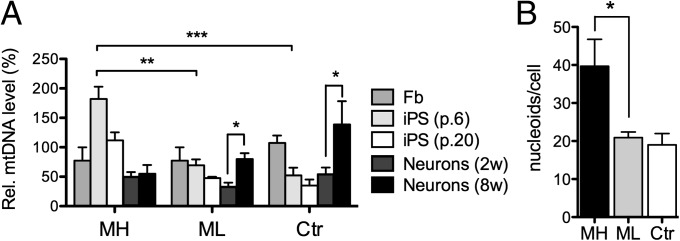
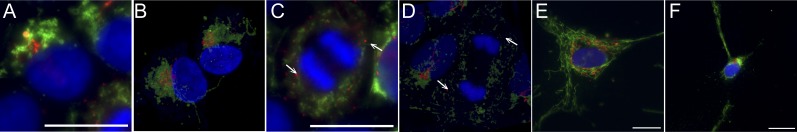
Mitochondrial DNA (mtDNA) mutations manifest with vast clinical heterogeneity. The molecular basis of this variability is mostly unknown because the lack of model systems has hampered mechanistic studies. We generated induced pluripotent stem cells from patients carrying the most common human disease mutation in mtDNA, m.3243A>G, underlying mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS) syndrome. During reprogramming, heteroplasmic mtDNA showed bimodal segregation toward homoplasmy, with concomitant changes in mtDNA organization, mimicking mtDNA bottleneck during epiblast specification. Induced pluripotent stem cell-derived neurons and various tissues derived from teratomas manifested cell-type specific respiratory chain (RC) deficiency patterns. Similar to MELAS patient tissues, complex I defect predominated. Upon neuronal differentiation, complex I specifically was sequestered in perinuclear PTEN-induced putative kinase 1 (PINK1) and Parkin-positive autophagosomes, suggesting active degradation through mitophagy. Other RC enzymes showed normal mitochondrial network distribution. Our data show that cellular context actively modifies RC deficiency manifestation in MELAS and that autophagy is a significant component of neuronal MELAS pathogenesis.
Keywords: disease modeling; mitochondria.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Skladal D, Halliday J, Thorburn DR. Minimum birth prevalence of mitochondrial respiratory chain disorders in children. Brain. 2003;126(Pt 8):1905–1912. - PubMed
-
- Holt IJ, Harding AE, Morgan-Hughes JA. Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature. 1988;331(6158):717–719. - PubMed
-
- Wallace DC, et al. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science. 1988;242(4884):1427–1430. - PubMed
-
- Ylikallio E, Suomalainen A. Mechanisms of mitochondrial diseases. Ann Med. 2012;44(1):41–59. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
