

A strategy to estimate unknown viral diversity in mammals
- PMID: 24003179
- PMCID: PMC3760253
- DOI: 10.1128/mBio.00598-13
A strategy to estimate unknown viral diversity in mammals
Abstract
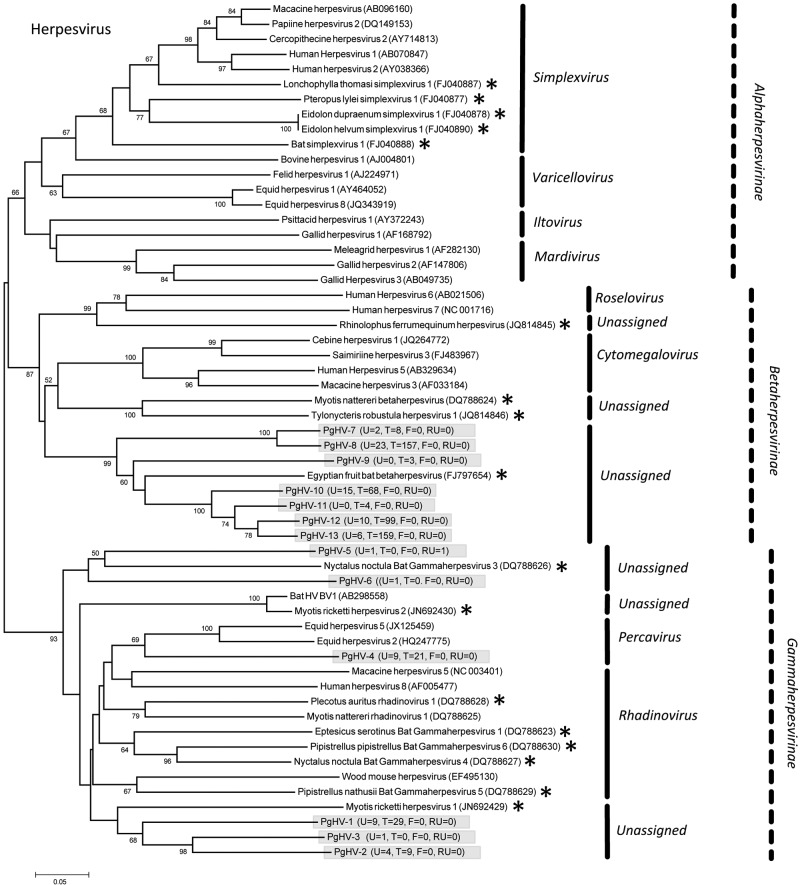
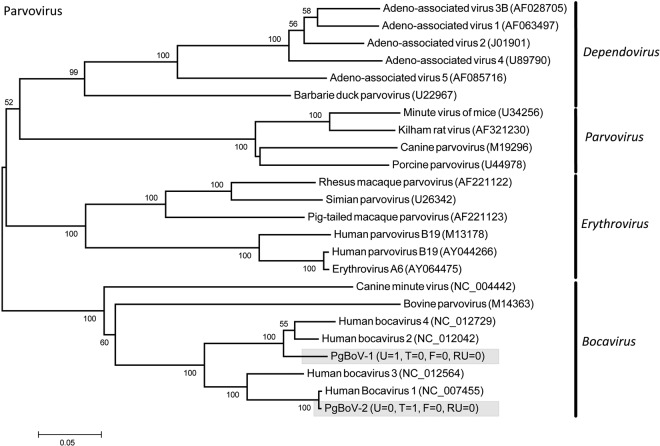
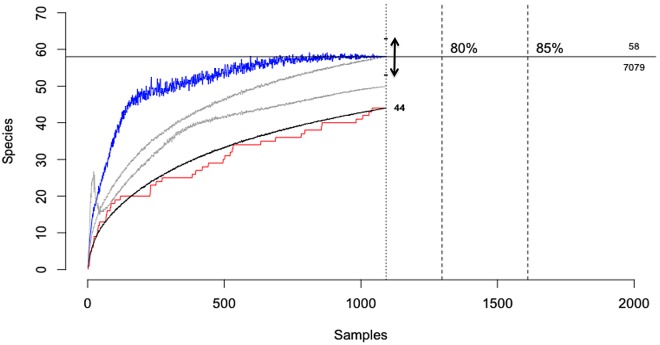
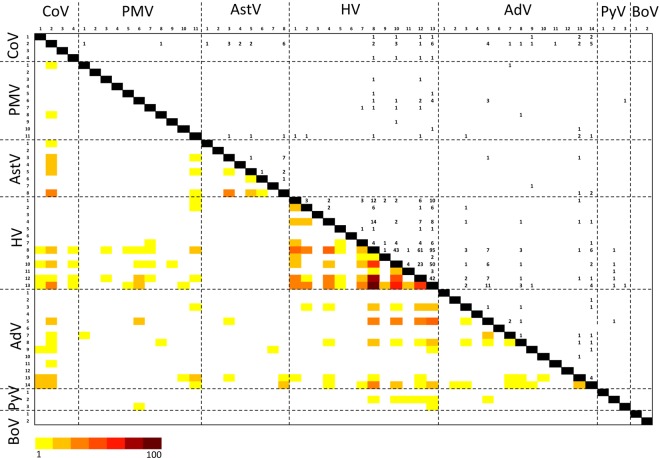
The majority of emerging zoonoses originate in wildlife, and many are caused by viruses. However, there are no rigorous estimates of total viral diversity (here termed "virodiversity") for any wildlife species, despite the utility of this to future surveillance and control of emerging zoonoses. In this case study, we repeatedly sampled a mammalian wildlife host known to harbor emerging zoonotic pathogens (the Indian Flying Fox, Pteropus giganteus) and used PCR with degenerate viral family-level primers to discover and analyze the occurrence patterns of 55 viruses from nine viral families. We then adapted statistical techniques used to estimate biodiversity in vertebrates and plants and estimated the total viral richness of these nine families in P. giganteus to be 58 viruses. Our analyses demonstrate proof-of-concept of a strategy for estimating viral richness and provide the first statistically supported estimate of the number of undiscovered viruses in a mammalian host. We used a simple extrapolation to estimate that there are a minimum of 320,000 mammalian viruses awaiting discovery within these nine families, assuming all species harbor a similar number of viruses, with minimal turnover between host species. We estimate the cost of discovering these viruses to be ~$6.3 billion (or ~$1.4 billion for 85% of the total diversity), which if annualized over a 10-year study time frame would represent a small fraction of the cost of many pandemic zoonoses.
Importance: Recent years have seen a dramatic increase in viral discovery efforts. However, most lack rigorous systematic design, which limits our ability to understand viral diversity and its ecological drivers and reduces their value to public health intervention. Here, we present a new framework for the discovery of novel viruses in wildlife and use it to make the first-ever estimate of the number of viruses that exist in a mammalian host. As pathogens continue to emerge from wildlife, this estimate allows us to put preliminary bounds around the potential size of the total zoonotic pool and facilitates a better understanding of where best to allocate resources for the subsequent discovery of global viral diversity.
Figures
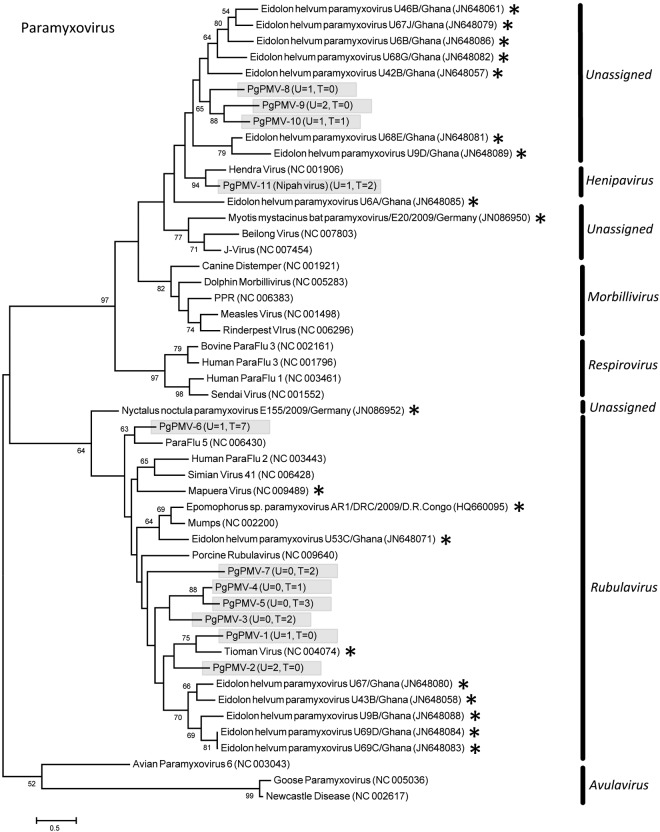
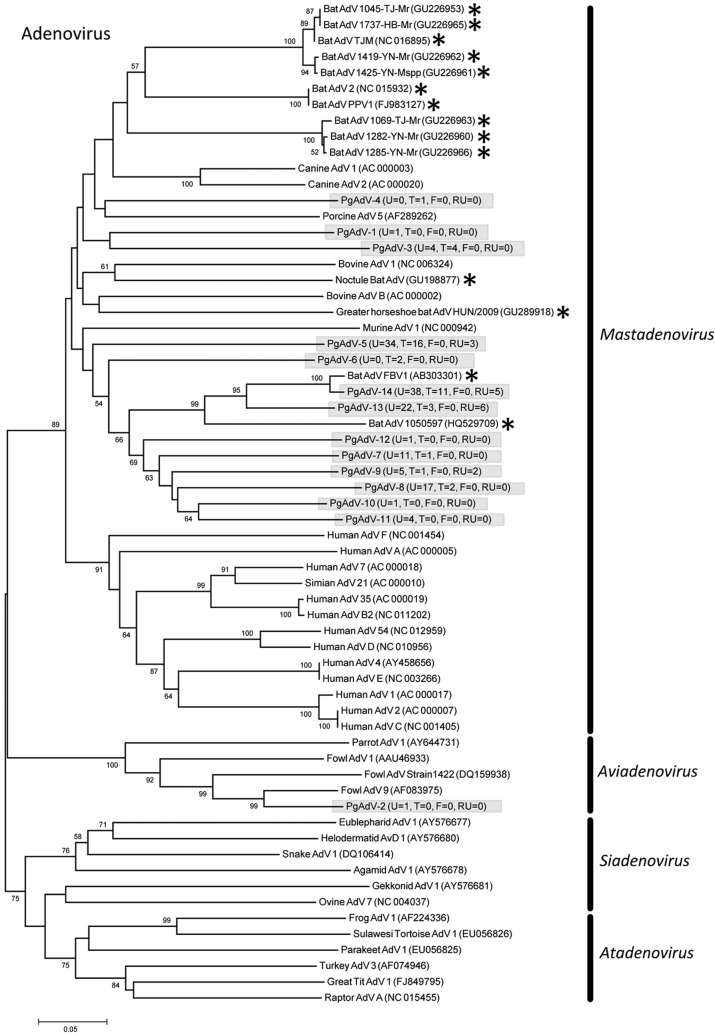

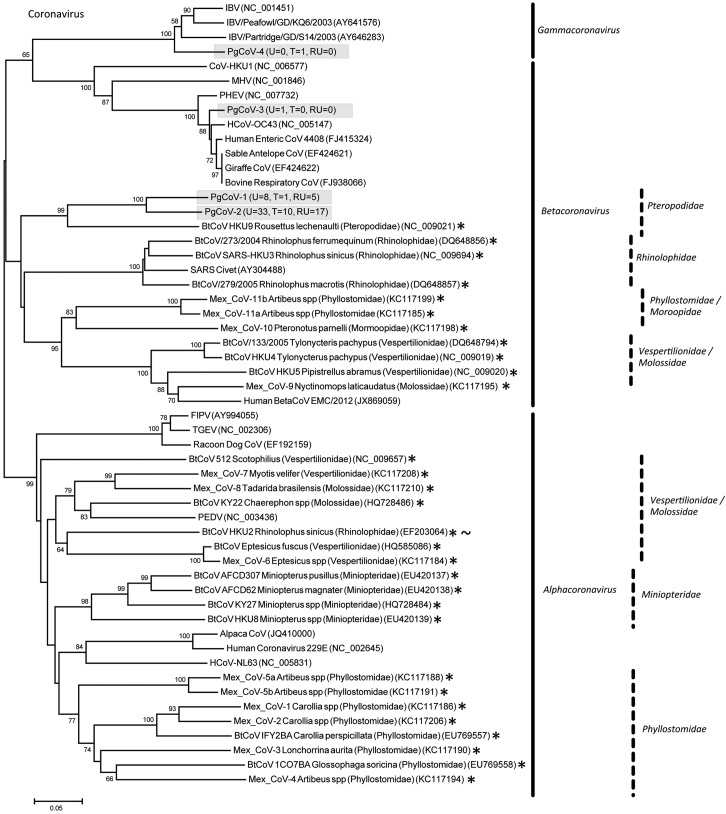
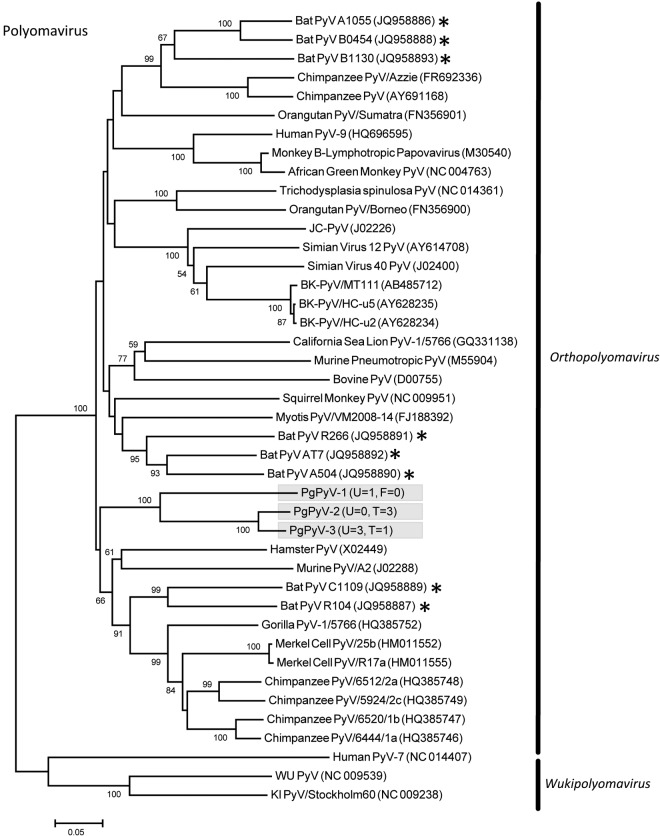
References
Publication types
MeSH terms
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
