

Transforming growth factor-β (TGF-β)-mediated connective tissue growth factor (CTGF) expression in hepatic stellate cells requires Stat3 signaling activation
- PMID: 24005672
- PMCID: PMC3798541
- DOI: 10.1074/jbc.M113.478685
Transforming growth factor-β (TGF-β)-mediated connective tissue growth factor (CTGF) expression in hepatic stellate cells requires Stat3 signaling activation
Abstract
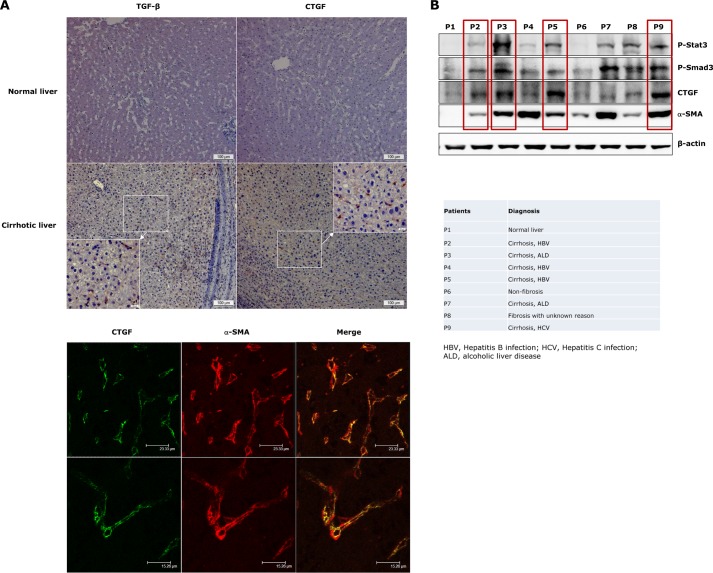
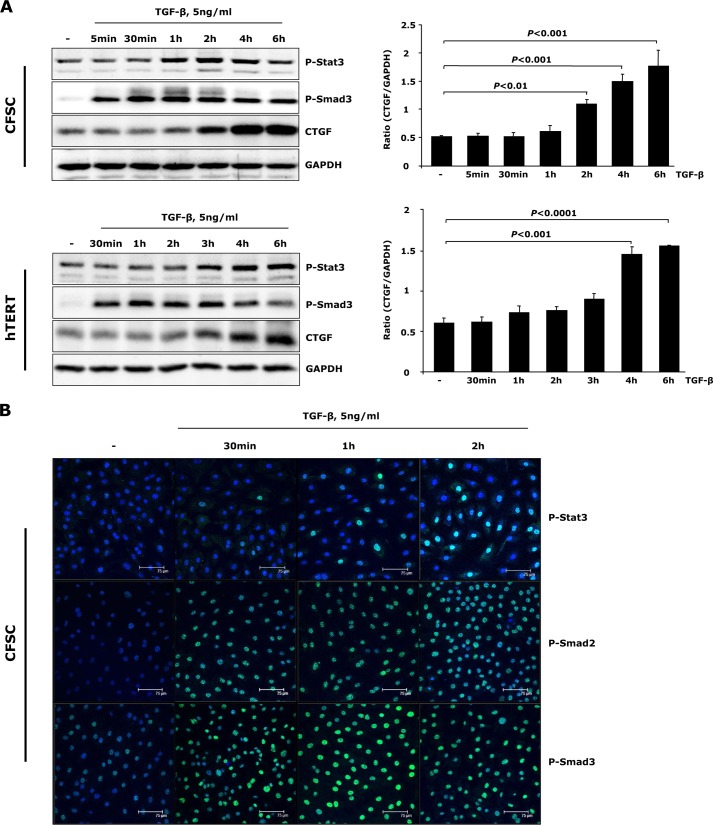
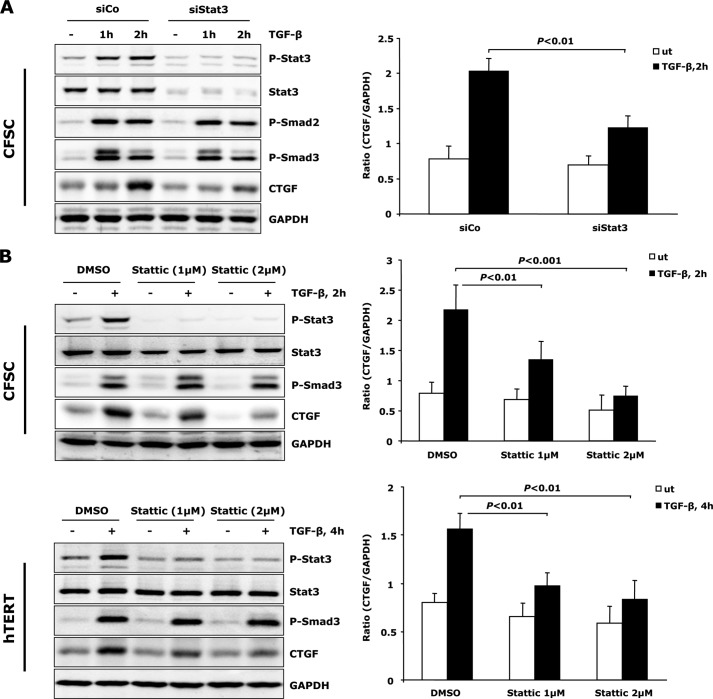
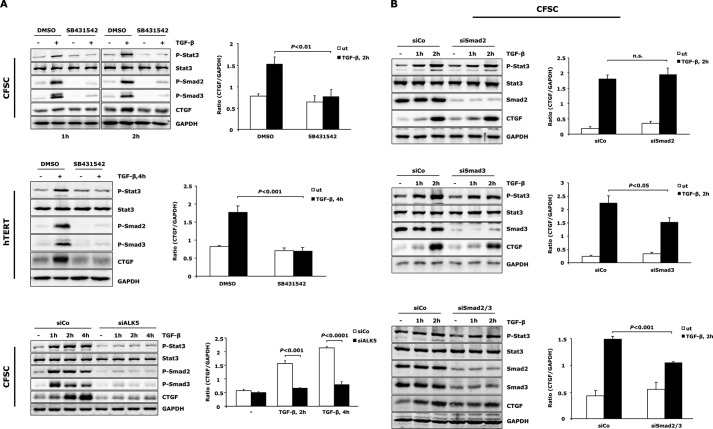
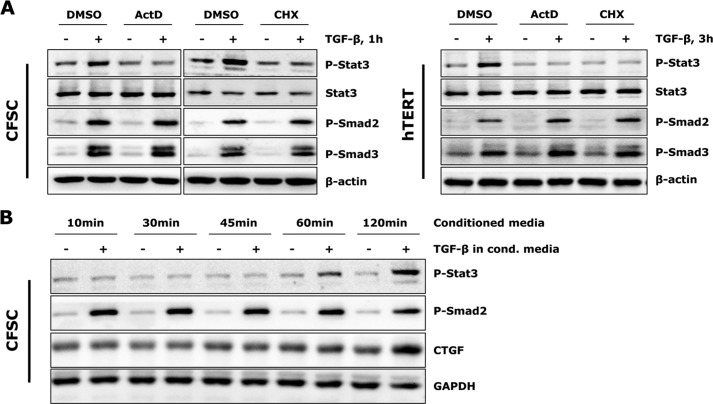
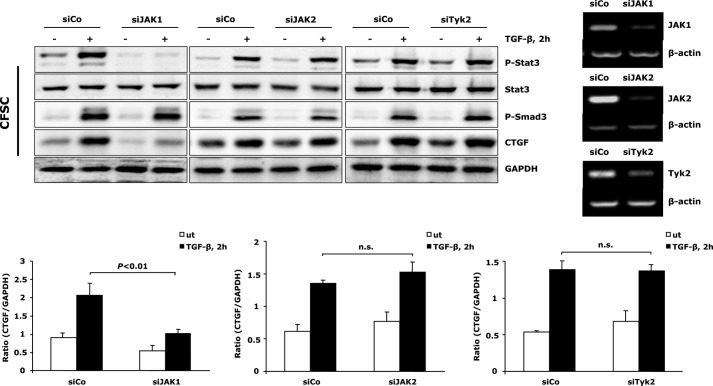
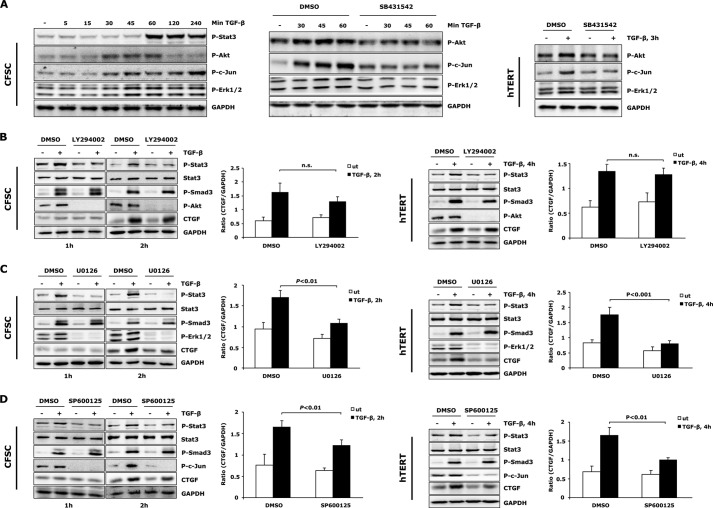
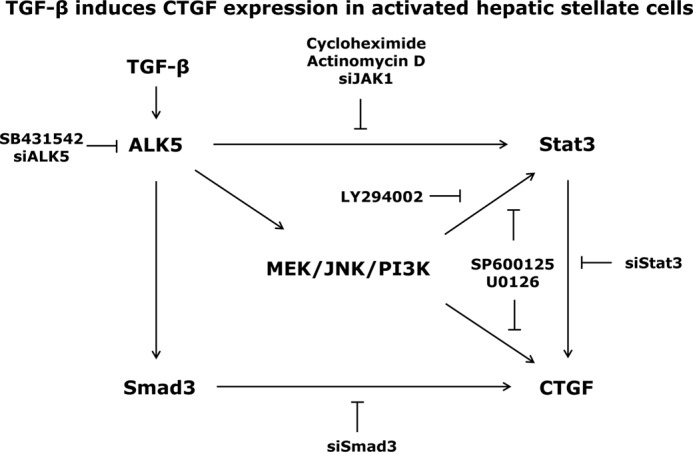
In fibrotic liver, connective tissue growth factor (CTGF) is constantly expressed in activated hepatic stellate cells (HSCs) and acts downstream of TGF-β to modulate extracellular matrix production. Distinct from other cell types in which Smad signaling plays major role in regulating CTGF production, TGF-β stimulated CTGF expression in activated HSCs is only in part dependent on Smad3. Other signaling molecules like MAPKs and PI3Ks may also participate in this process, and the underlying mechanisms have yet to be clarified. In this study, we report involvement of Stat3 activation in modulating CTGF production upon TGF-β challenge in activated HSCs. Stat3 is phosphorylated via JAK1 and acts as a critical ALK5 (activin receptor-like kinase 5) downstream signaling molecule to mediate CTGF expression. This process requires de novo gene transcription and is additionally modulated by MEK1/2, JNK, and PI3K pathways. Cell-specific knockdown of Smad3 partially decreases CTGF production, whereas it has no significant influence on Stat3 activation. The total CTGF production induced by TGF-β in activated HSCs is therefore, to a large extent, dependent on the balance and integration of the canonical Smad3 and Stat3 signaling pathways.
Keywords: ERK; Jak Kinase; Jun N-terminal Kinase (JNK); Liver Fibrosis; MAP Kinases (MAPKs); PI 3-kinase (PI3K); SMAD Transcription Factor.
Figures
References
-
- Leask A., Abraham D. J. (2003) The role of connective tissue growth factor, a multifunctional matricellular protein, in fibroblast biology. Biochem. Cell Biol. 81, 355–363 - PubMed
-
- Gressner O. A., Gressner A. M. (2008) Connective tissue growth factor: a fibrogenic master switch in fibrotic liver diseases. Liver Int. 28, 1065–1079 - PubMed
-
- Williams E. J., Gaça M. D., Brigstock D. R., Arthur M. J., Benyon R. C. (2000) Increased expression of connective tissue growth factor in fibrotic human liver and in activated hepatic stellate cells. J. Hepatol. 32, 754–761 - PubMed
-
- Abou-Shady M., Friess H., Zimmermann A., di Mola F. F., Guo X. Z., Baer H. U., Büchler M. W. (2000) Connective tissue growth factor in human liver cirrhosis. Liver 20, 296–304 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
