

Epigenetic mechanisms of neurodegeneration in Huntington's disease
- PMID: 24006238
- PMCID: PMC3805871
- DOI: 10.1007/s13311-013-0206-5
Epigenetic mechanisms of neurodegeneration in Huntington's disease
Abstract
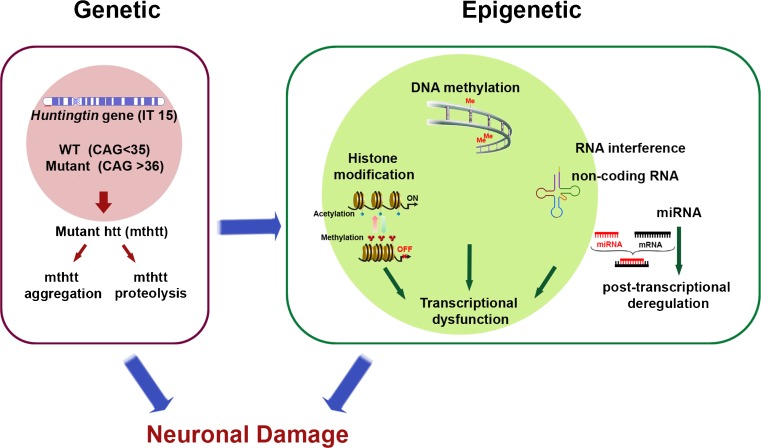
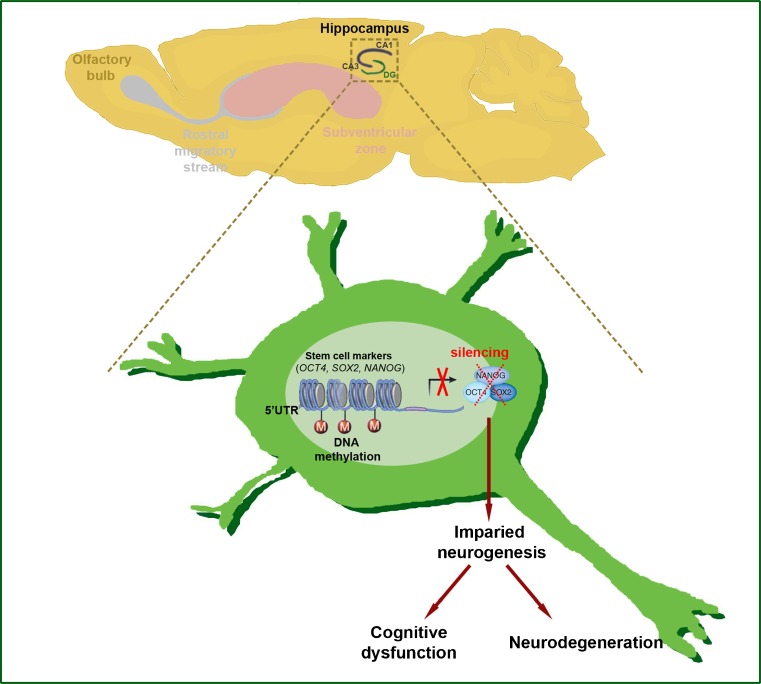
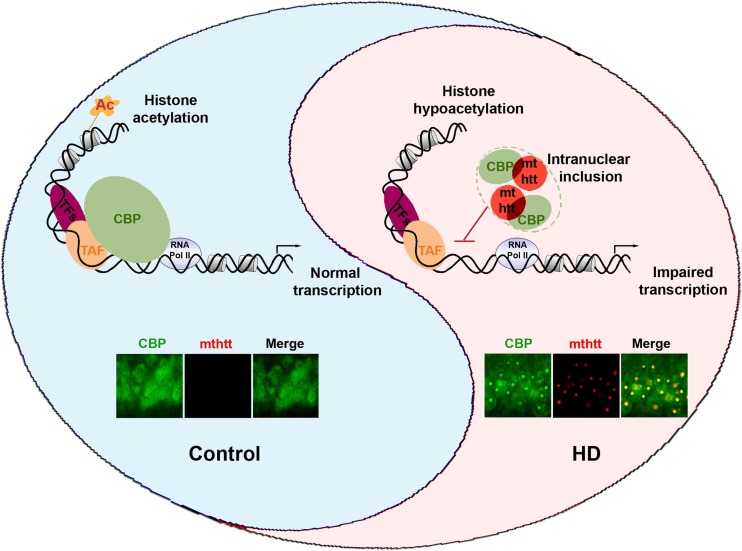
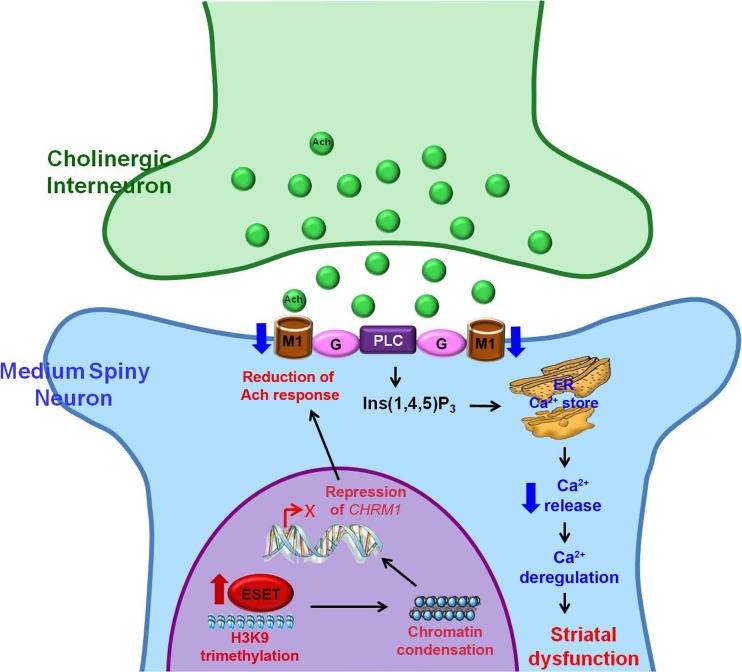
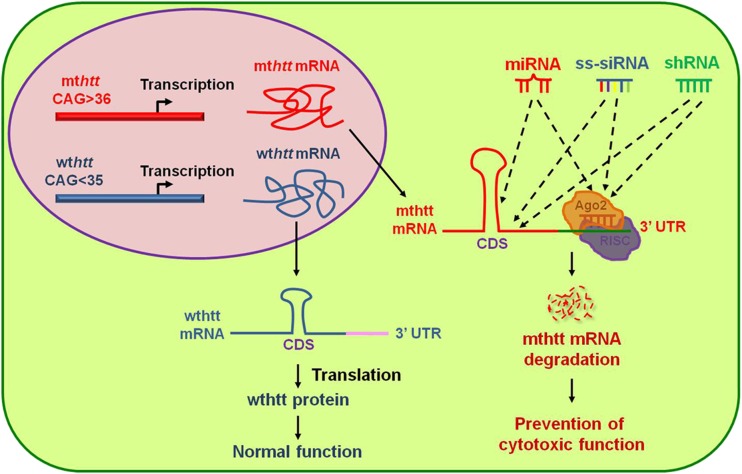
Huntington's disease (HD) is an incurable and fatal hereditary neurodegenerative disorder of mid-life onset characterized by chorea, emotional distress, and progressive cognitive decline. HD is caused by an expansion of CAG repeats coding for glutamine (Q) in exon 1 of the huntingtin gene. Recent studies suggest that epigenetic modifications may play a key role in HD pathogenesis. Alterations of the epigenetic "histone code" lead to chromatin remodeling and deregulation of neuronal gene transcription that are prominently linked to HD pathogenesis. Furthermore, specific noncoding RNAs and microRNAs are associated with neuronal damage in HD. In this review, we discuss how DNA methylation, post-translational modifications of histone, and noncoding RNA function are affected and involved in HD pathogenesis. In addition, we summarize the therapeutic effects of histone deacetylase inhibitors and DNA binding drugs on epigenetic modifications and neuropathological sequelae in HD. Our understanding of the role of these epigenetic mechanisms may lead to the identification of novel biological markers and new therapeutic targets to treat HD.
Figures
References
-
- Myers RH, MacDonald ME, Koroshetz WJ, et al. De novo expansion of a (CAG)n repeat in sporadic Huntington’s disease. Nat Genet. 1993;5:168–173. - PubMed
-
- Kremer BP, Goldberg S, Andrew J, et al. A worldwide study of the Huntington’s disease mutation the sensitivity and specificity of measuring CAG repeats. New Engl J Med. 1994;330:1401–1406. - PubMed
-
- Huntington G. On chorea. Med Surg Rep (Philadelphia) 1872;317–321.
-
- Jergelsma G. Nue anatomische befunde bei paralysis agitans und bei chronischer progressive chorea. Neurol Centralbl. 1908;27:995–996.
-
- Bruyn GW, Bots GTAM, Dom R. Huntinton’s chorea: Current neuropathological status. In: Chase TN, Wexler NS, Barbeau A, editors. Huntington’s disease. Advances in neurology. New York: Raven Press; 1979. pp. 83–93.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
