

Assessing the utility of coevolution-based residue-residue contact predictions in a sequence- and structure-rich era
- PMID: 24009338
- PMCID: PMC3785744
- DOI: 10.1073/pnas.1314045110
Assessing the utility of coevolution-based residue-residue contact predictions in a sequence- and structure-rich era
Erratum in
- Proc Natl Acad Sci U S A. 2013 Nov 12;110(46):18734
Abstract
Recently developed methods have shown considerable promise in predicting residue-residue contacts in protein 3D structures using evolutionary covariance information. However, these methods require large numbers of evolutionarily related sequences to robustly assess the extent of residue covariation, and the larger the protein family, the more likely that contact information is unnecessary because a reasonable model can be built based on the structure of a homolog. Here we describe a method that integrates sequence coevolution and structural context information using a pseudolikelihood approach, allowing more accurate contact predictions from fewer homologous sequences. We rigorously assess the utility of predicted contacts for protein structure prediction using large and representative sequence and structure databases from recent structure prediction experiments. We find that contact predictions are likely to be accurate when the number of aligned sequences (with sequence redundancy reduced to 90%) is greater than five times the length of the protein, and that accurate predictions are likely to be useful for structure modeling if the aligned sequences are more similar to the protein of interest than to the closest homolog of known structure. These conditions are currently met by 422 of the protein families collected in the Pfam database.
Keywords: markov random field; maximum-entropy model; protein coevolution.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
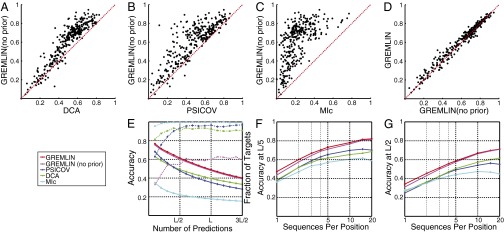
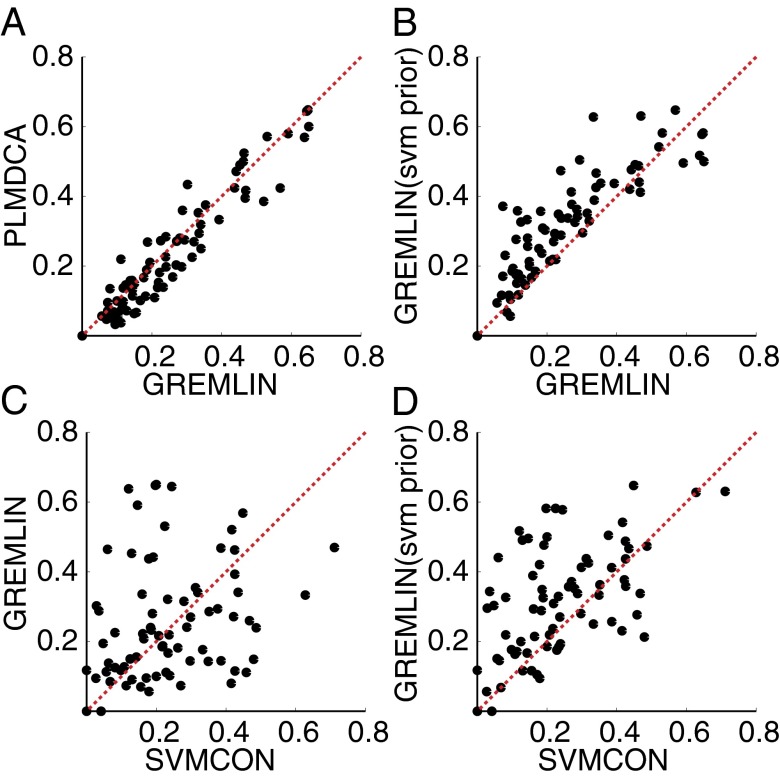
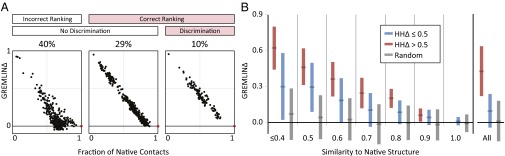
 predicts GREMLINΔ: GREMLINΔ versus structural similarity of homolog to native structure computed by TM-align (14) (for homologs of all targets with high-resolution crystal structures < 2.1 Å). When
predicts GREMLINΔ: GREMLINΔ versus structural similarity of homolog to native structure computed by TM-align (14) (for homologs of all targets with high-resolution crystal structures < 2.1 Å). When  (blue bars), GREMLINΔ is rarely better than random (green bars, constructed by pooling 100 permutations of predicted scores for each target). When
(blue bars), GREMLINΔ is rarely better than random (green bars, constructed by pooling 100 permutations of predicted scores for each target). When  (red bars), GREMLINΔ is significantly positive and contact scores successfully discriminate between native and homology model even when the homolog is likely to be from the same fold (similarity
(red bars), GREMLINΔ is significantly positive and contact scores successfully discriminate between native and homology model even when the homolog is likely to be from the same fold (similarity  ). Error bars show mean and SD of distributions in all cases.
). Error bars show mean and SD of distributions in all cases.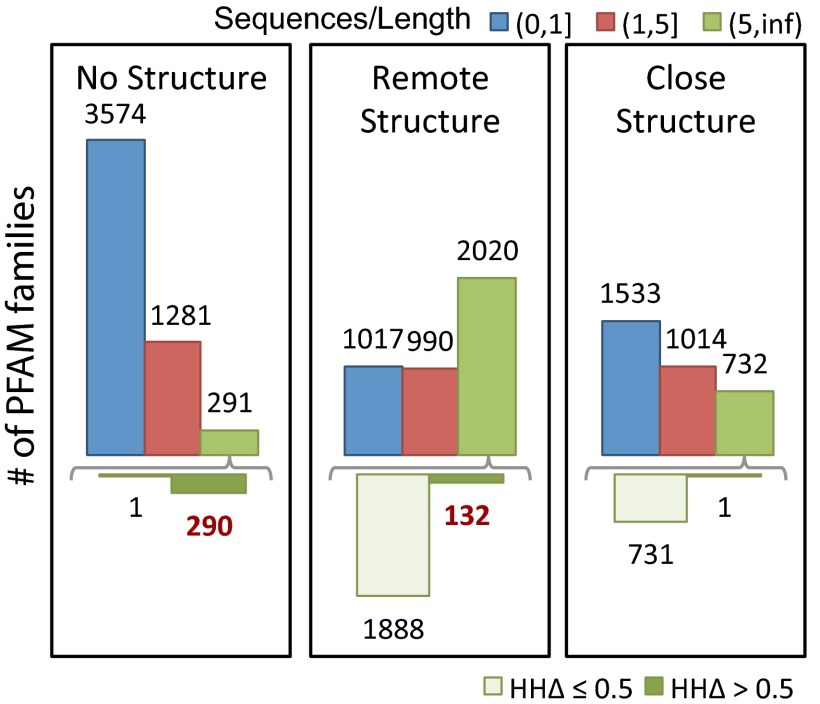
 to the closest protein of known structure is shown in the lower panel. In cases where the difference in profiles is large (
to the closest protein of known structure is shown in the lower panel. In cases where the difference in profiles is large ( : right bar in each group, Lower), these predictions are likely to improve on comparative models.
: right bar in each group, Lower), these predictions are likely to improve on comparative models.References
-
- Tress ML, Valencia A. Predicted residue–residue contacts can help the scoring of 3d models. Proteins. Struct Funct Bioinf. 2010;78(8):1980–1991. - PubMed
-
- Jones DT, Buchan DWA, Cozzetto D, Pontil M. PSICOV: Precise structural contact prediction using sparse inverse covariance estimation on large multiple sequence alignments. Bioinformatics. 2012;28(2):184–190. - PubMed
-
- Balakrishnan S, Kamisetty H, Carbonell JG, Lee SI, Langmead CJ. Learning generative models for protein fold families. Protiens Struct Funct Bioinf. 2011;79(4):1061–1078. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
