

A Markovian analysis of bacterial genome sequence constraints
- PMID: 24010012
- PMCID: PMC3757466
- DOI: 10.7717/peerj.127
A Markovian analysis of bacterial genome sequence constraints
Abstract
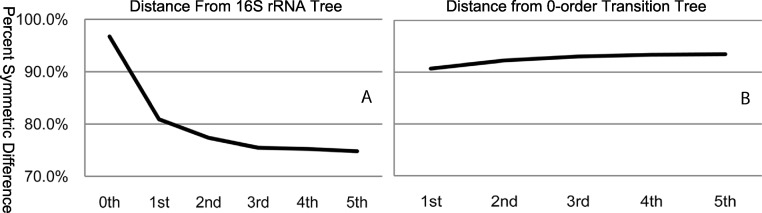
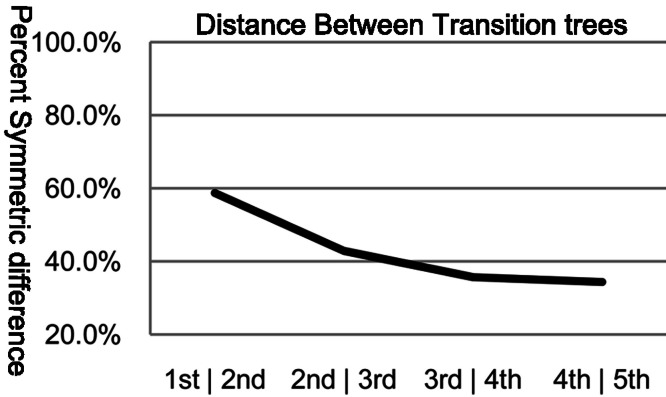
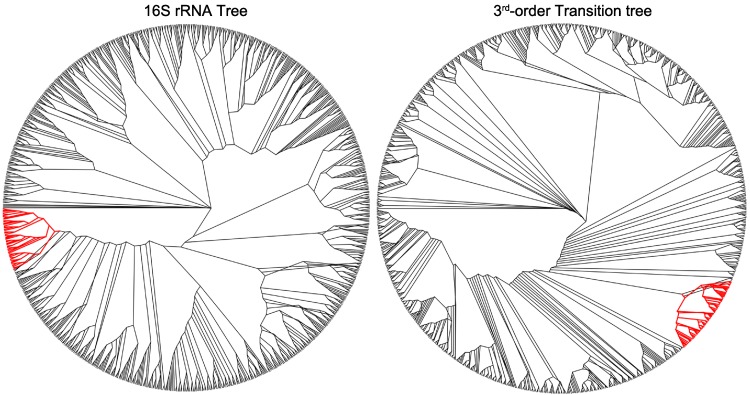
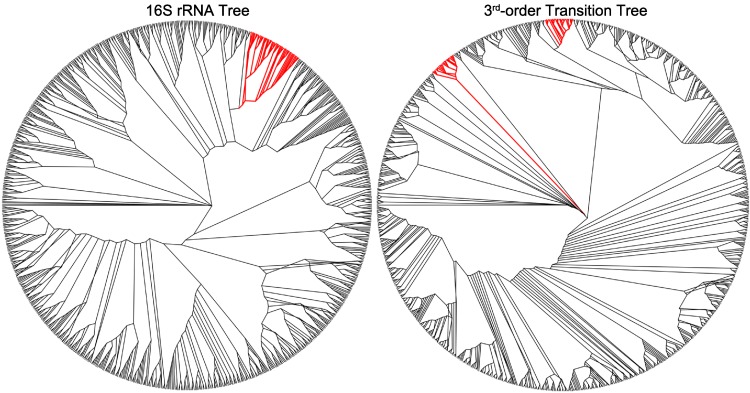
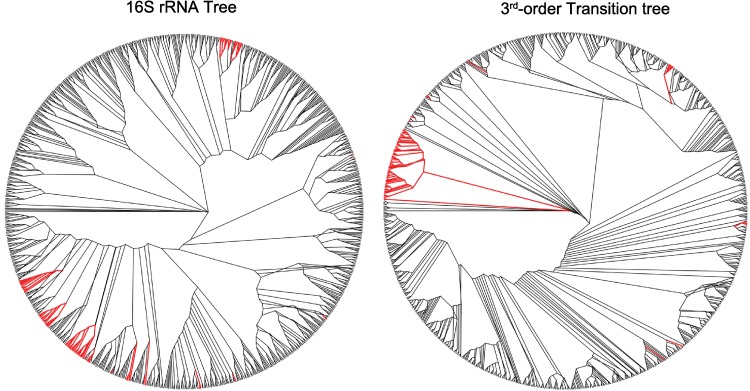
The arrangement of nucleotides within a bacterial chromosome is influenced by numerous factors. The degeneracy of the third codon within each reading frame allows some flexibility of nucleotide selection; however, the third nucleotide in the triplet of each codon is at least partly determined by the preceding two. This is most evident in organisms with a strong G + C bias, as the degenerate codon must contribute disproportionately to maintaining that bias. Therefore, a correlation exists between the first two nucleotides and the third in all open reading frames. If the arrangement of nucleotides in a bacterial chromosome is represented as a Markov process, we would expect that the correlation would be completely captured by a second-order Markov model and an increase in the order of the model (e.g., third-, fourth-…order) would not capture any additional uncertainty in the process. In this manuscript, we present the results of a comprehensive study of the Markov property that exists in the DNA sequences of 906 bacterial chromosomes. All of the 906 bacterial chromosomes studied exhibit a statistically significant Markov property that extends beyond second-order, and therefore cannot be fully explained by codon usage. An unrooted tree containing all 906 bacterial chromosomes based on their transition probability matrices of third-order shares ∼25% similarity to a tree based on sequence homologies of 16S rRNA sequences. This congruence to the 16S rRNA tree is greater than for trees based on lower-order models (e.g., second-order), and higher-order models result in diminishing improvements in congruence. A nucleotide correlation most likely exists within every bacterial chromosome that extends past three nucleotides. This correlation places significant limits on the number of nucleotide sequences that can represent probable bacterial chromosomes. Transition matrix usage is largely conserved by taxa, indicating that this property is likely inherited, however some important exceptions exist that may indicate the convergent evolution of some bacteria.
Keywords: Bacteria; Markov model; Sequencing; Topology; rRNA.
Figures
Similar articles
-
Constraint on di-nucleotides by codon usage bias in bacterial genomes.Gene. 2014 Feb 15;536(1):18-28. doi: 10.1016/j.gene.2013.11.098. Epub 2013 Dec 11. Gene. 2014. PMID: 24333347
-
Bacterial genomes lacking long-range correlations may not be modeled by low-order Markov chains: the role of mixing statistics and frame shift of neighboring genes.Comput Biol Chem. 2014 Dec;53 Pt A:15-25. doi: 10.1016/j.compbiolchem.2014.08.005. Epub 2014 Aug 30. Comput Biol Chem. 2014. PMID: 25257406
-
Natural selection retains overrepresented out-of-frame stop codons against frameshift peptides in prokaryotes.BMC Genomics. 2010 Sep 9;11:491. doi: 10.1186/1471-2164-11-491. BMC Genomics. 2010. PMID: 20828396 Free PMC article.
-
A stochastic analysis of three viral sequences.Mol Biol Evol. 1992 Jul;9(4):666-77. doi: 10.1093/oxfordjournals.molbev.a040741. Mol Biol Evol. 1992. PMID: 1321321
-
Evolutionary implications of microbial genome tetranucleotide frequency biases.Genome Res. 2003 Feb;13(2):145-58. doi: 10.1101/gr.335003. Genome Res. 2003. PMID: 12566393 Free PMC article.
Cited by
-
Quantitative Analysis of Axonal Branch Dynamics in the Developing Nervous System.PLoS Comput Biol. 2016 Mar 21;12(3):e1004813. doi: 10.1371/journal.pcbi.1004813. eCollection 2016 Mar. PLoS Comput Biol. 2016. PMID: 26998842 Free PMC article.
-
Multi-AI competing and winning against humans in iterated Rock-Paper-Scissors game.Sci Rep. 2020 Aug 17;10(1):13873. doi: 10.1038/s41598-020-70544-7. Sci Rep. 2020. PMID: 32807813 Free PMC article.
-
Automatic block-wise genotype-phenotype association detection based on hidden Markov model.BMC Bioinformatics. 2023 Apr 7;24(1):138. doi: 10.1186/s12859-023-05265-5. BMC Bioinformatics. 2023. PMID: 37029361 Free PMC article.
References
-
- Achtman M, Zurth K, Morelli G, Torrea G, Guiyoule A, Carniel E. Yersinia pestis, the cause of plague, is a recently emerged clone of Yersinia pseudotuberculosis. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:14043–14048. doi: 10.1073/pnas.96.24.14043. - DOI - PMC - PubMed
-
- Anderson TW, Goodman LA. Statistical-inference about Markov-chains. Annals of Mathematical Statistics. 1957;28:89–110. doi: 10.1214/aoms/1177707039. - DOI
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
