

Protein modeling: what happened to the "protein structure gap"?
- PMID: 24010712
- PMCID: PMC3816506
- DOI: 10.1016/j.str.2013.08.007
Protein modeling: what happened to the "protein structure gap"?
Abstract
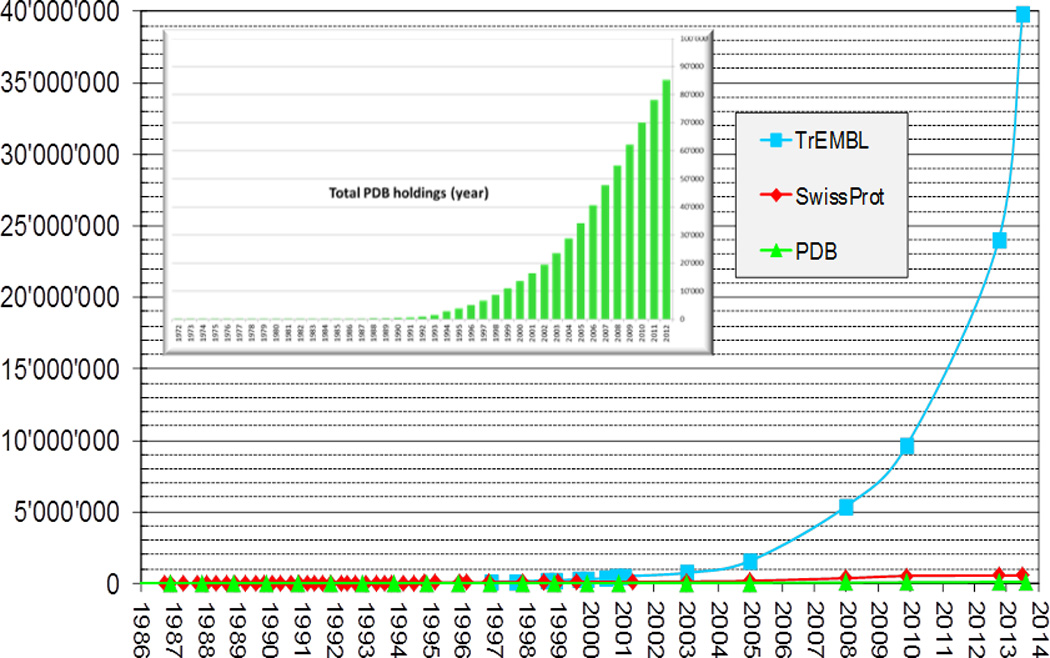
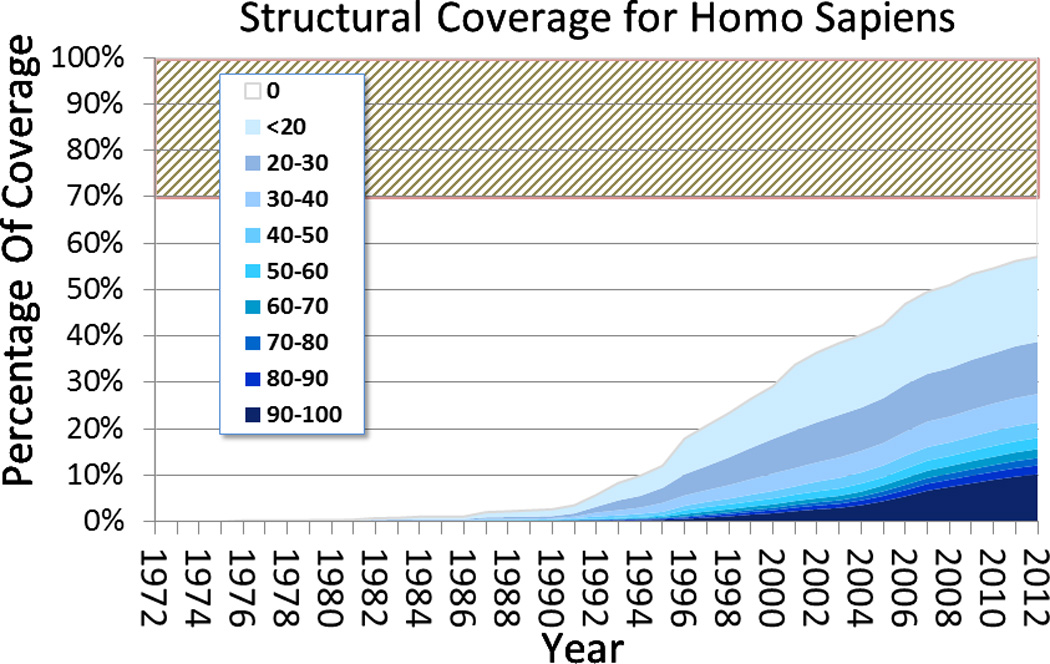
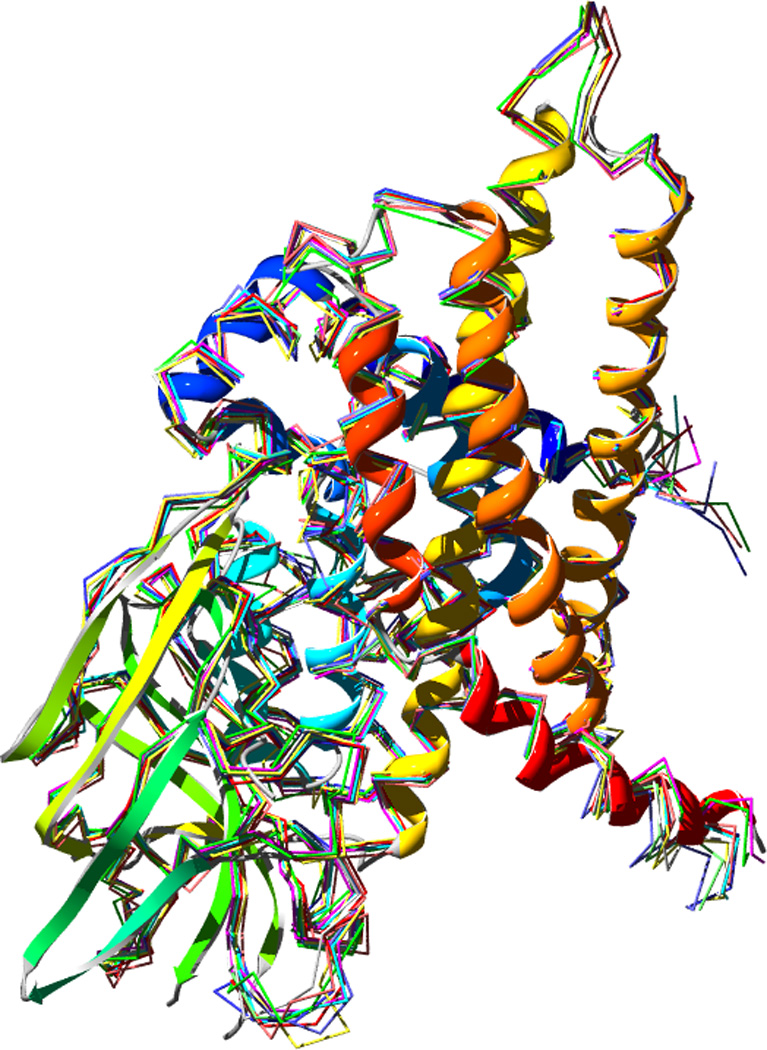
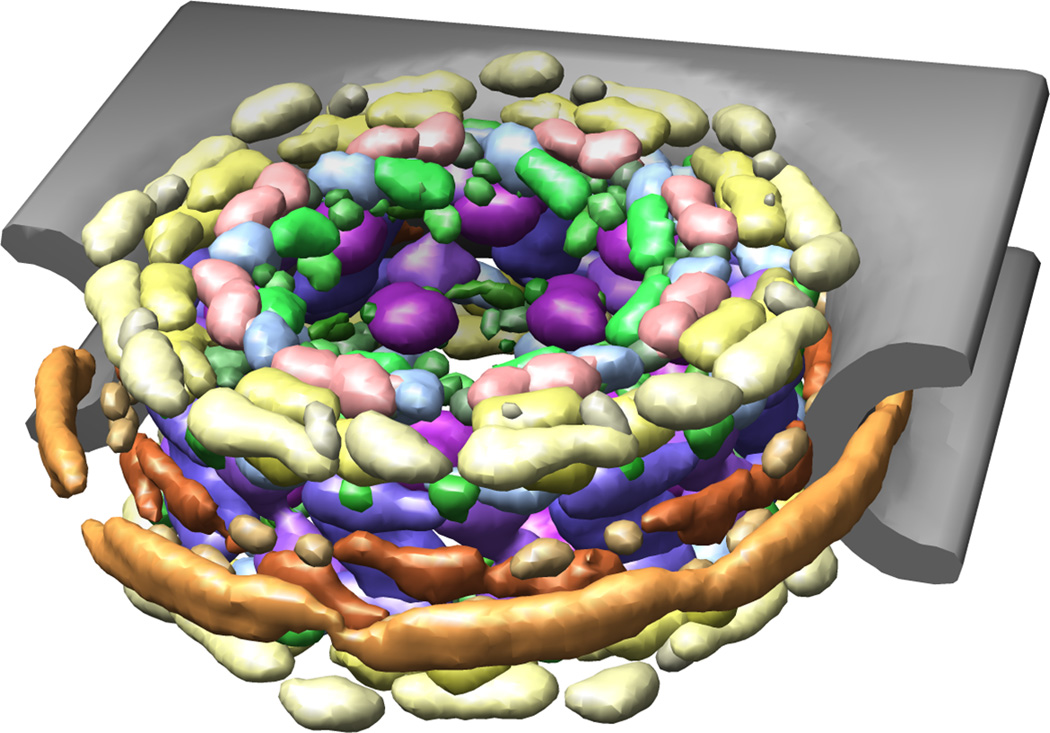
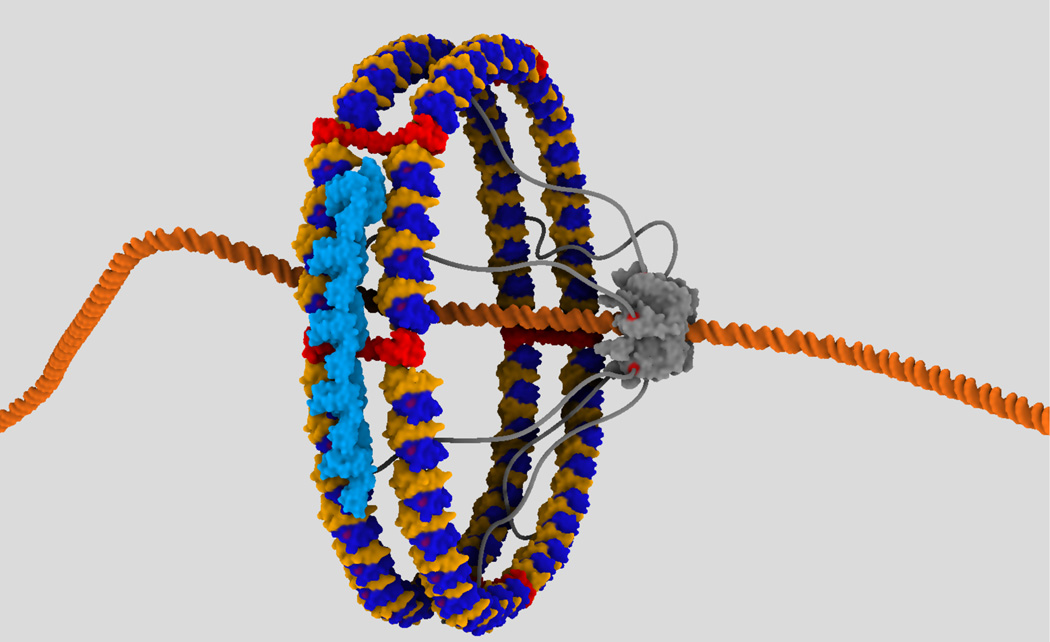
Computational modeling of three-dimensional macromolecular structures and complexes from their sequence has been a long-standing vision in structural biology. Over the last 2 decades, a paradigm shift has occurred: starting from a large "structure knowledge gap" between the huge number of protein sequences and small number of known structures, today, some form of structural information, either experimental or template-based models, is available for the majority of amino acids encoded by common model organism genomes. With the scientific focus of interest moving toward larger macromolecular complexes and dynamic networks of interactions, the integration of computational modeling methods with low-resolution experimental techniques allows the study of large and complex molecular machines. One of the open challenges for computational modeling and prediction techniques is to convey the underlying assumptions, as well as the expected accuracy and structural variability of a specific model, which is crucial to understanding its limitations.
Copyright © 2013 Elsevier Ltd. All rights reserved.
Figures

References
-
- Alber F, Dokudovskaya S, Veenhoff LM, Zhang W, Kipper J, Devos D, Suprapto A, Karni-Schmidt O, Williams R, Chait BT, et al. Determining the architectures of macromolecular assemblies. Nature. 2007a;450:683–694. - PubMed
-
- Alber F, Dokudovskaya S, Veenhoff LM, Zhang W, Kipper J, Devos D, Suprapto A, Karni-Schmidt O, Williams R, Chait BT, et al. The molecular architecture of the nuclear pore complex. Nature. 2007b;450:695–701. - PubMed
-
- Alber F, Forster F, Korkin D, Topf M, Sali A. Integrating diverse data for structure determination of macromolecular assemblies. Annu Rev Biochem. 2008;77:443–477. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
