

NLRP3 inflammasome blockade inhibits VEGF-A-induced age-related macular degeneration
- PMID: 24012762
- PMCID: PMC3821550
- DOI: 10.1016/j.celrep.2013.08.002
NLRP3 inflammasome blockade inhibits VEGF-A-induced age-related macular degeneration
Abstract
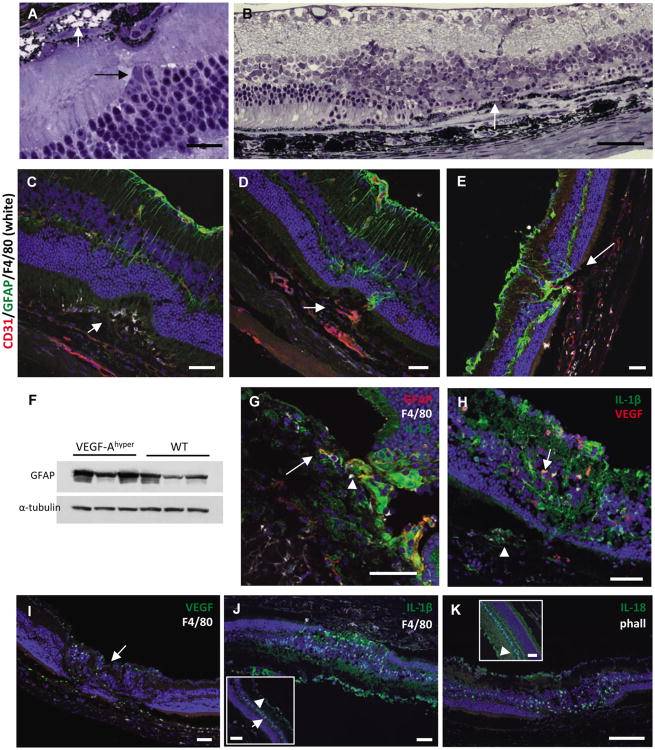
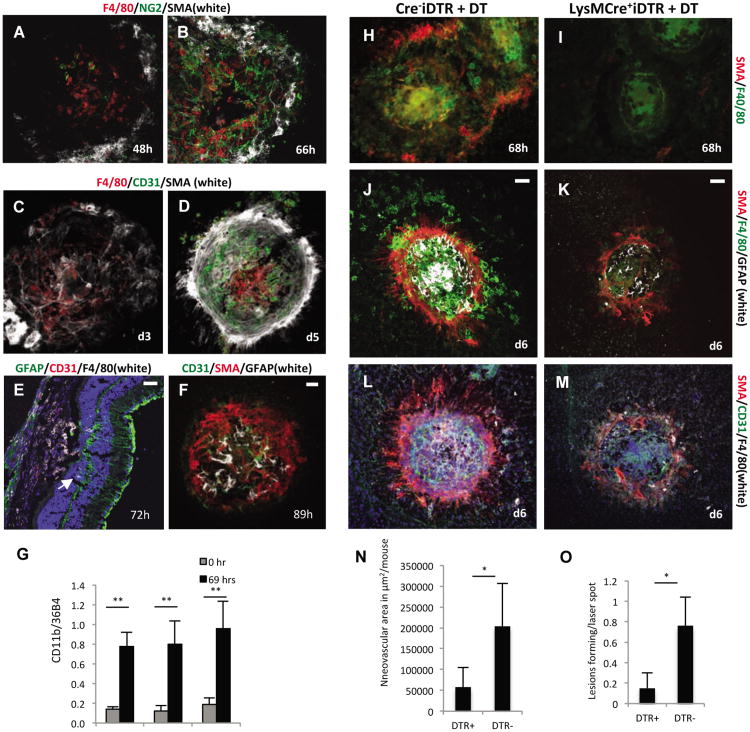
The NLRP3 inflammasome is activated in age-related macular degeneration (AMD), but it remains unknown whether its activation contributes to AMD pathologies. VEGF-A is increased in neovascular ("wet") AMD, but it is not known whether it plays a role in inflammasome activation, whether an increase of VEGF-A by itself is sufficient to cause neovascular AMD and whether it can contribute to nonexudative ("dry") AMD that often co-occurs with the neovascular form. Here, it is shown that an increase in VEGF-A results in NLRP3 inflammasome activation and is sufficient to cause both forms of AMD pathologies. Targeting NLRP3 or the inflammasome effector cytokine IL-1β inhibits but does not prevent VEGF-A-induced AMD pathologies, whereas targeting IL-18 promotes AMD. Thus, increased VEGF-A provides a unifying pathomechanism for both forms of AMD; combining therapeutic inhibition of both VEGF-A and IL-1β or the NLRP3 inflammasome is therefore likely to suppress both forms of AMD.
Copyright © 2013 The Author. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
No conflict of interest is reported.
Figures
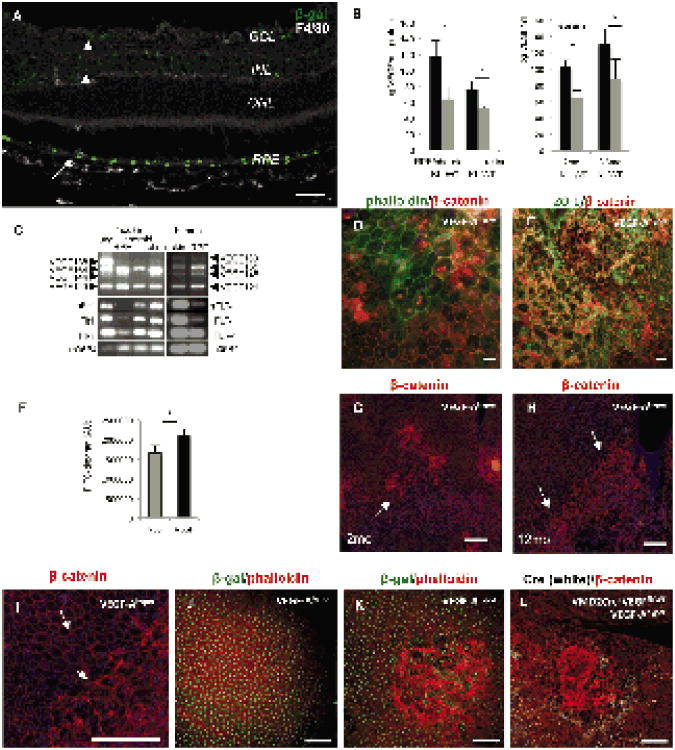
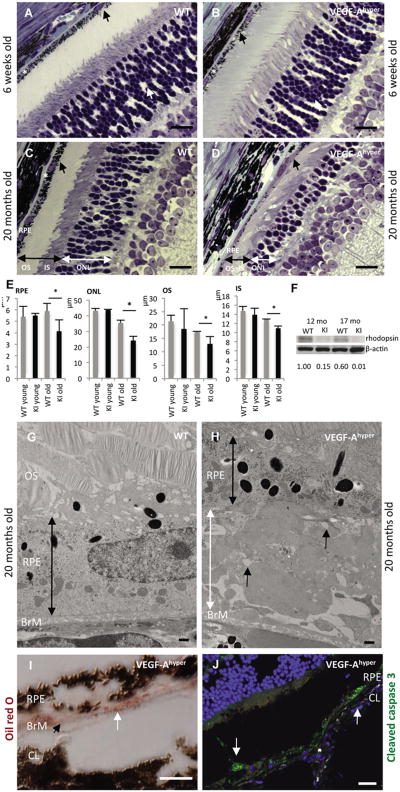
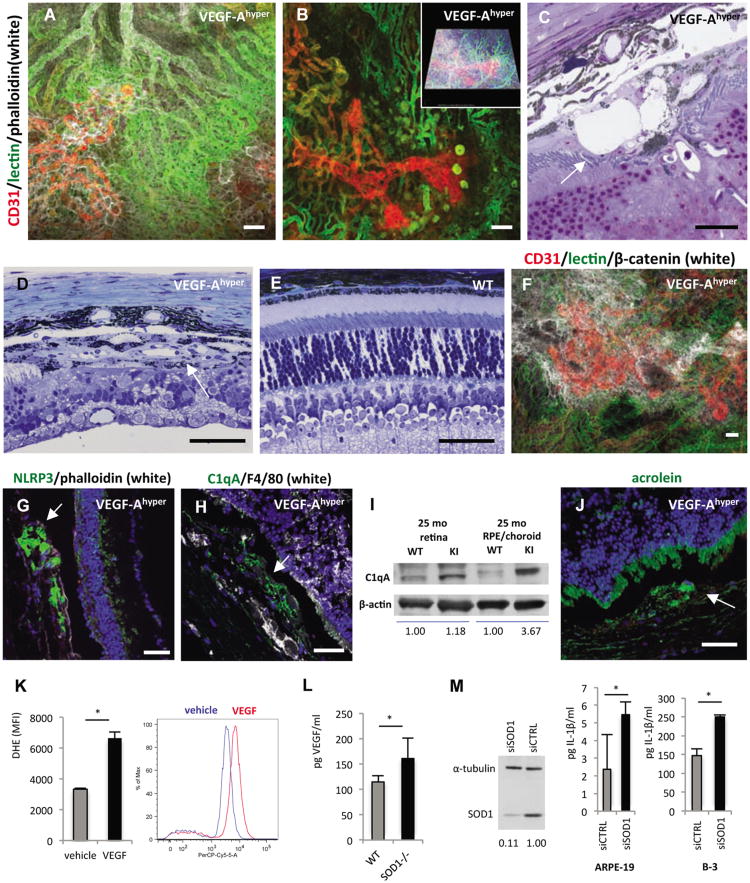
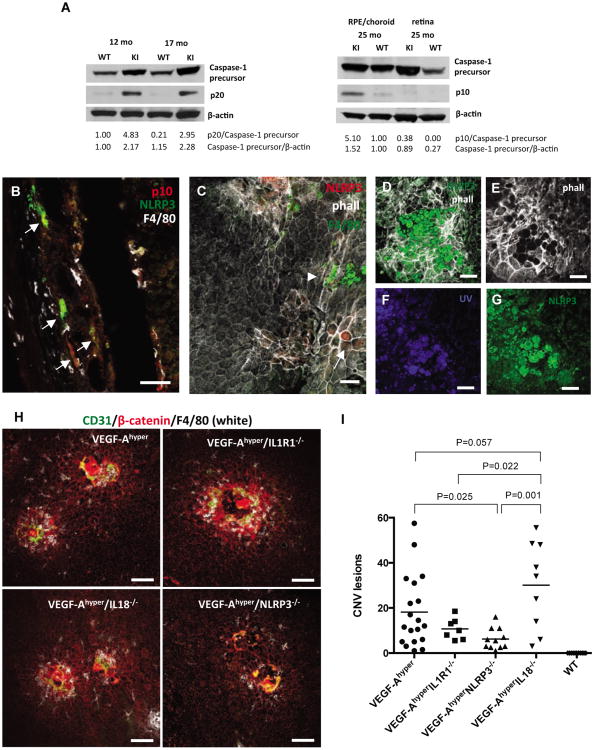
References
-
- Anderson DH, Mullins RF, Hageman GS, Johnson LV. A role for local inflammation in the formation of drusen in the aging eye. American journal of ophthalmology. 2002;134:411–431. - PubMed
-
- Bird AC, Bressler NM, Bressler SB, Chisholm IH, Coscas G, Davis MD, de Jong PT, Klaver CC, Klein BE, Klein R, et al. An international classification and grading system for age-related maculopathy and age-related macular degeneration. The International ARM Epidemiological Study Group. Surv Ophthalmol. 1995;39:367–374. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
