

Measurement and modeling of transcriptional noise in the cell cycle regulatory network
- PMID: 24013422
- PMCID: PMC3865016
- DOI: 10.4161/cc.26257
Measurement and modeling of transcriptional noise in the cell cycle regulatory network
Abstract
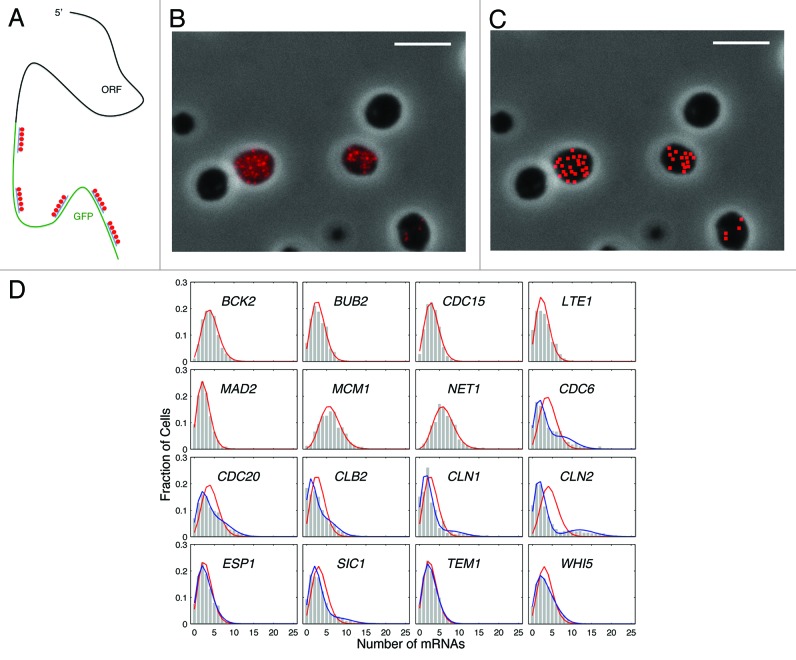
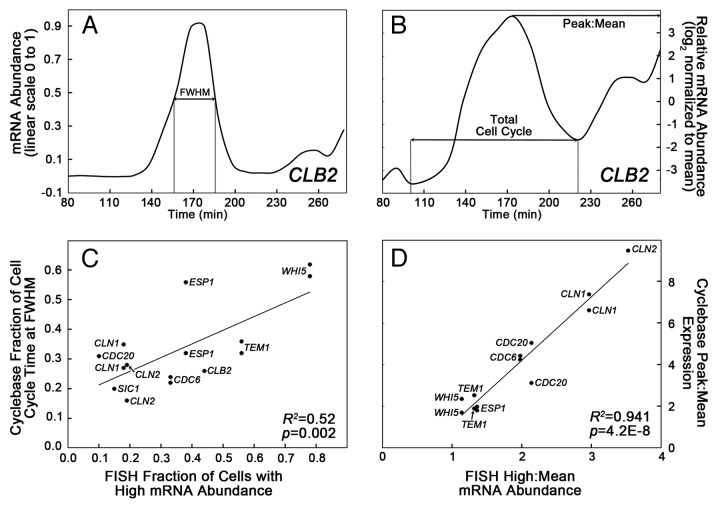
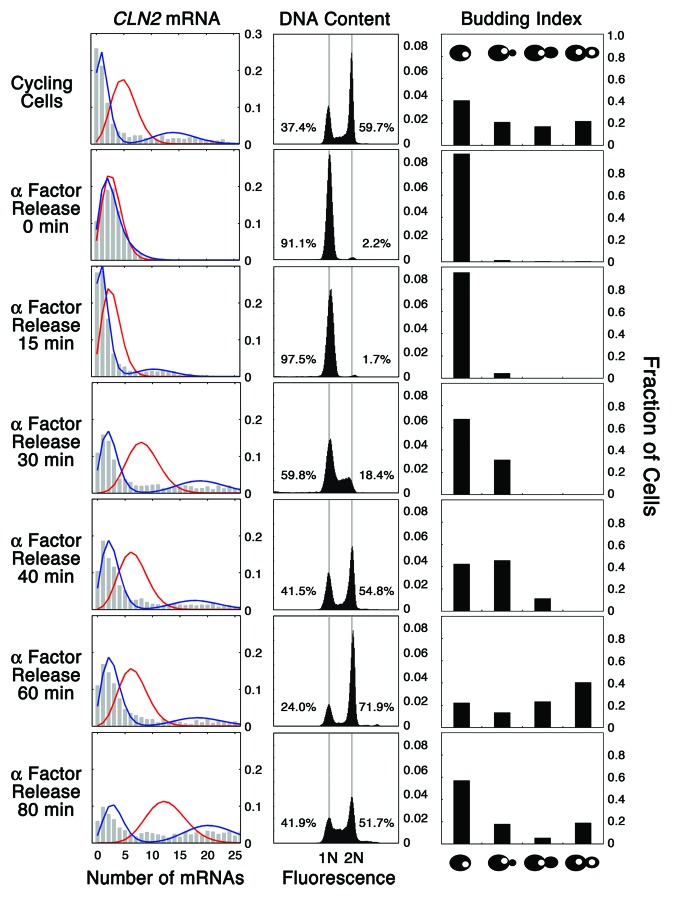
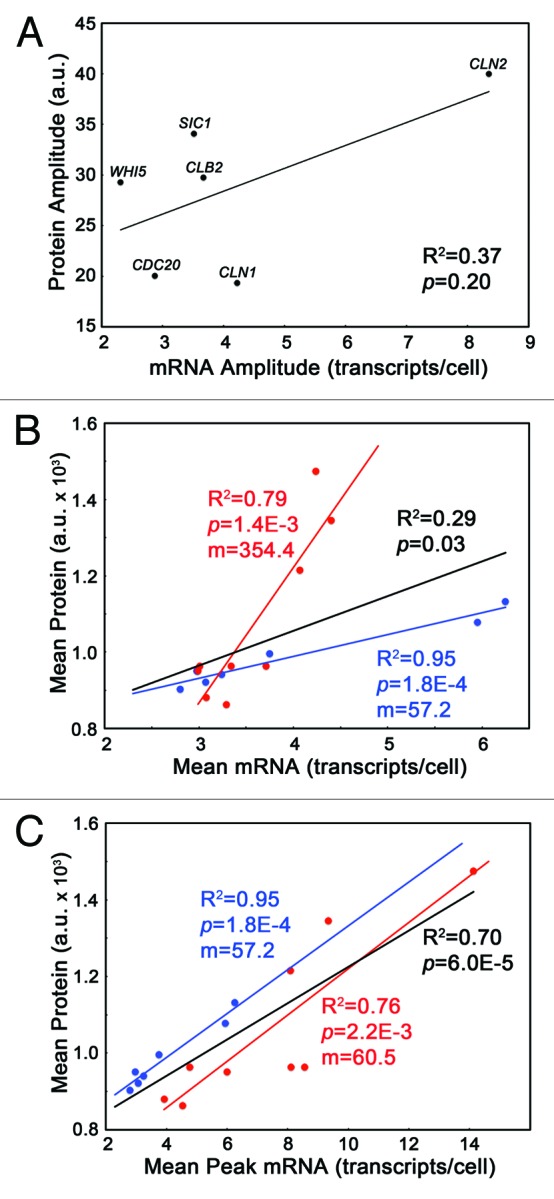
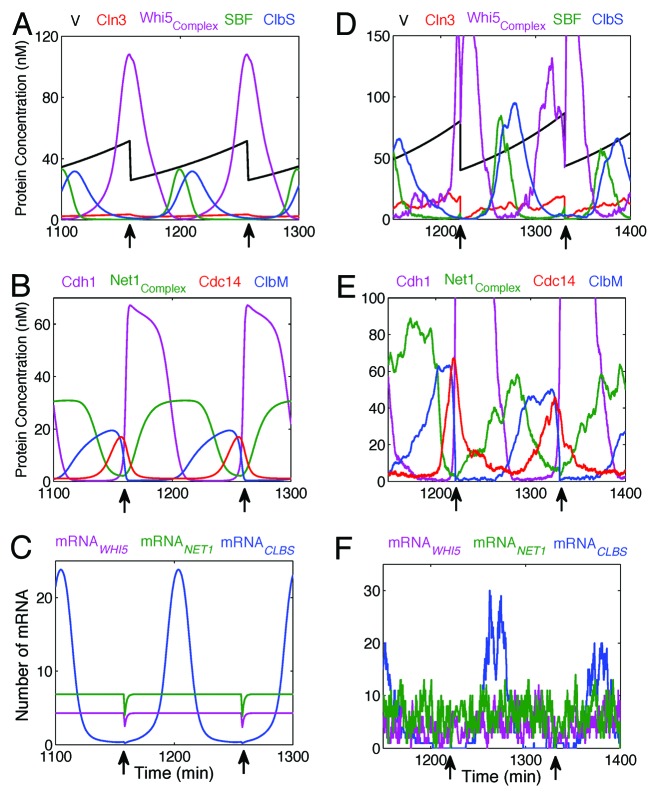
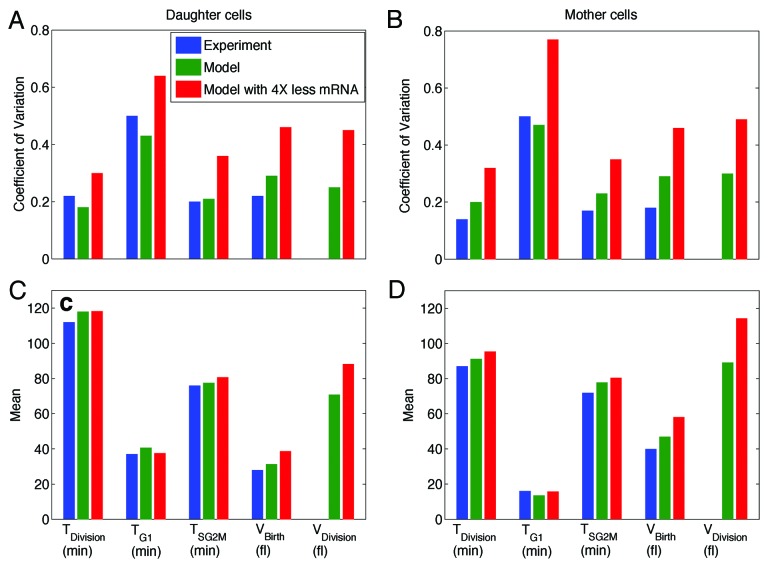
Fifty years of genetic and molecular experiments have revealed a wealth of molecular interactions involved in the control of cell division. In light of the complexity of this control system, mathematical modeling has proved useful in analyzing biochemical hypotheses that can be tested experimentally. Stochastic modeling has been especially useful in understanding the intrinsic variability of cell cycle events, but stochastic modeling has been hampered by a lack of reliable data on the absolute numbers of mRNA molecules per cell for cell cycle control genes. To fill this void, we used fluorescence in situ hybridization (FISH) to collect single molecule mRNA data for 16 cell cycle regulators in budding yeast, Saccharomyces cerevisiae. From statistical distributions of single-cell mRNA counts, we are able to extract the periodicity, timing, and magnitude of transcript abundance during the cell cycle. We used these parameters to improve a stochastic model of the cell cycle to better reflect the variability of molecular and phenotypic data on cell cycle progression in budding yeast.
Keywords: Saccharomycescerevisiae; cell cycle; gene expression noise; single mRNA FISH; stochastic modeling.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases