

Nuclear quantum effects and hydrogen bond fluctuations in water
- PMID: 24014589
- PMCID: PMC3785726
- DOI: 10.1073/pnas.1308560110
Nuclear quantum effects and hydrogen bond fluctuations in water
Abstract
The hydrogen bond (HB) is central to our understanding of the properties of water. However, despite intense theoretical and experimental study, it continues to hold some surprises. Here, we show from an analysis of ab initio simulations that take proper account of nuclear quantum effects that the hydrogen-bonded protons in liquid water experience significant excursions in the direction of the acceptor oxygen atoms. This generates a small but nonnegligible fraction of transient autoprotolysis events that are not seen in simulations with classical nuclei. These events are associated with major rearrangements of the electronic density, as revealed by an analysis of the computed Wannier centers and (1)H chemical shifts. We also show that the quantum fluctuations exhibit significant correlations across neighboring HBs, consistent with an ephemeral shuttling of protons along water wires. We end by suggesting possible implications for our understanding of how perturbations (solvated ions, interfaces, and confinement) might affect the HB network in water.
Keywords: ab initio liquid water; generalized Langevin equation thermostat; path integral molecular dynamics.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
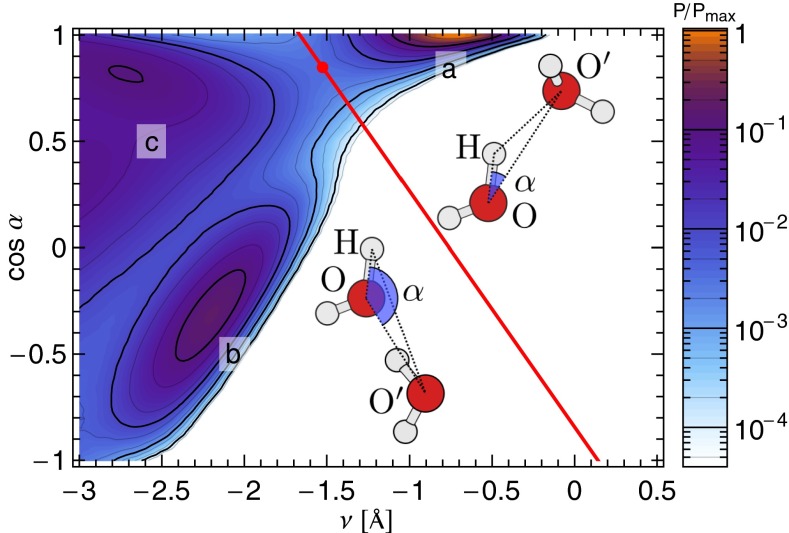
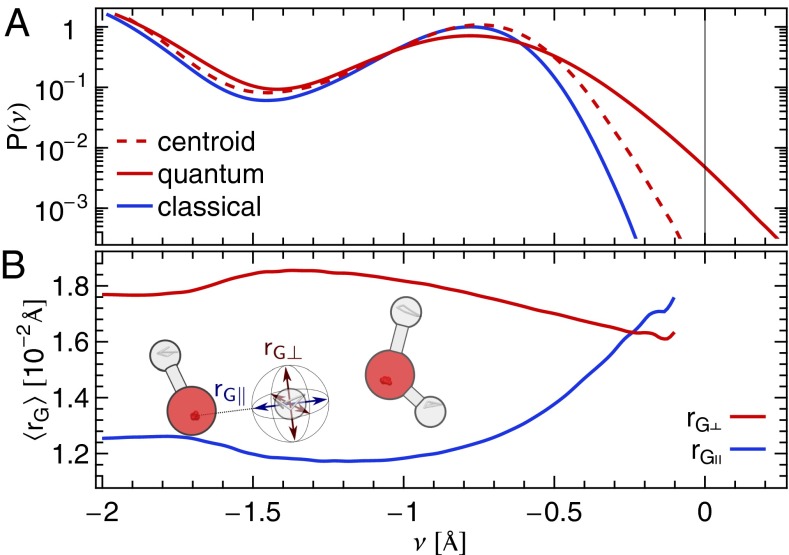
 and perpendicular
and perpendicular  to the O–H covalent bond for different values of ν for the centroid.
to the O–H covalent bond for different values of ν for the centroid.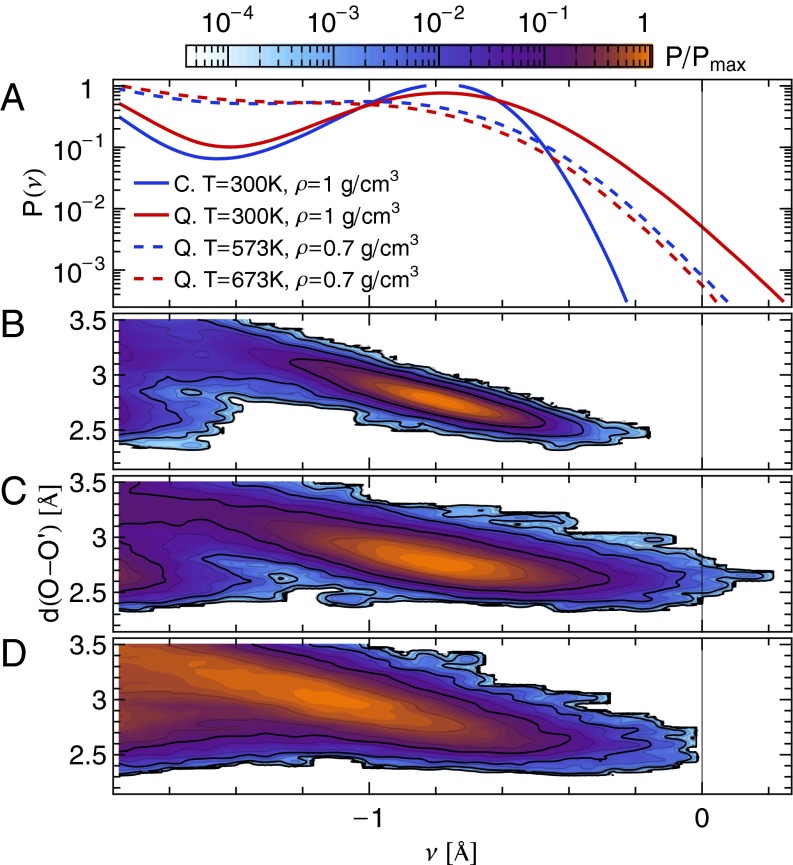
 ,
,  ; (C) quantum nuclei,
; (C) quantum nuclei,  ,
,  ; and (D) quantum nuclei,
; and (D) quantum nuclei,  ,
,  .
.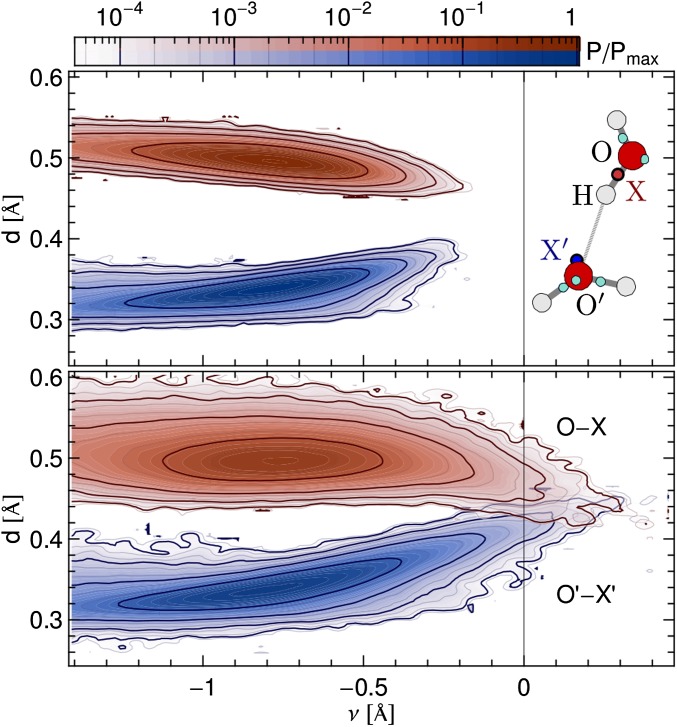
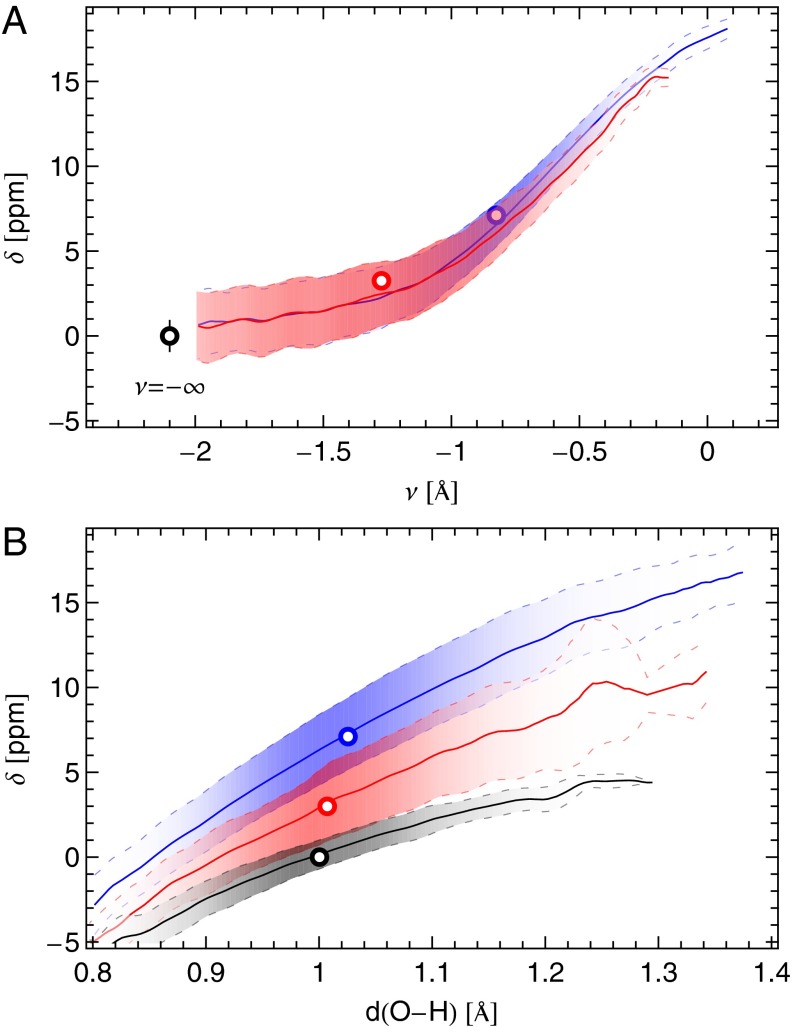
 and
and  (blue), supercritical water at
(blue), supercritical water at  and
and  (red), and an isolated gas-phase water molecule at 473 K (black). In all cases, NQEs were included using PIGLET. The continuous lines correspond to conditional averages of the chemical shift as a function of the structural parameter, the dashed lines delimit the area within 1 standard deviation from the mean, and the shading is proportional to the probability distribution of the structural parameter. The dots indicate the means of the structural parameter and the chemical shift. (A) Proton-transfer coordinate ν is used as the structural parameter, the gas-phase molecule corresponding to an asymptotic value of
(red), and an isolated gas-phase water molecule at 473 K (black). In all cases, NQEs were included using PIGLET. The continuous lines correspond to conditional averages of the chemical shift as a function of the structural parameter, the dashed lines delimit the area within 1 standard deviation from the mean, and the shading is proportional to the probability distribution of the structural parameter. The dots indicate the means of the structural parameter and the chemical shift. (A) Proton-transfer coordinate ν is used as the structural parameter, the gas-phase molecule corresponding to an asymptotic value of  . (B) Covalent bond length
. (B) Covalent bond length  is used.
is used.References
-
- Eisenberg D, Kauzmann W (1968) The Structure and Properties of Water (Oxford Univ Press, Oxford)
-
- Pauling L (1960) The Nature of the Chemical Bond and the Structure of Molecules and Crystals: An Introduction to Modern Structural Chemistry (Cornell Univ Press, Ithaca, NY), Vol 18.
-
- Silvestrelli PL, Parrinello M. Structural, electronic, and bonding properties of liquid water from first principles. J Chem Phys. 1999;111(8):3572–3580.
-
- Sharma M, Resta R, Car R. Intermolecular dynamical charge fluctuations in water: A signature of the H-bond network. Phys Rev Lett. 2005;95(18):187401. - PubMed
-
- Boero M, Terakura K, Ikeshoji T, Liew CC, Parrinello M. Hydrogen bonding and dipole moment of water at supercritical conditions: A first-principles molecular dynamics study. Phys Rev Lett. 2000;85(15):3245–3248. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
