

Tyrosinemia type 1: a rare and forgotten cause of reversible hypertrophic cardiomyopathy in infancy
- PMID: 24016420
- PMCID: PMC3846631
- DOI: 10.1186/1756-0500-6-362
Tyrosinemia type 1: a rare and forgotten cause of reversible hypertrophic cardiomyopathy in infancy
Abstract
Background: Tyrosinemia type 1 (TT1) is an autosomal recessive disorder caused by deficiency of the enzyme fumarylacetoacetate hydrolase (FAH). TT1 usually presents in infancy with features suggestive of liver disease or with sepsis-like symptoms.
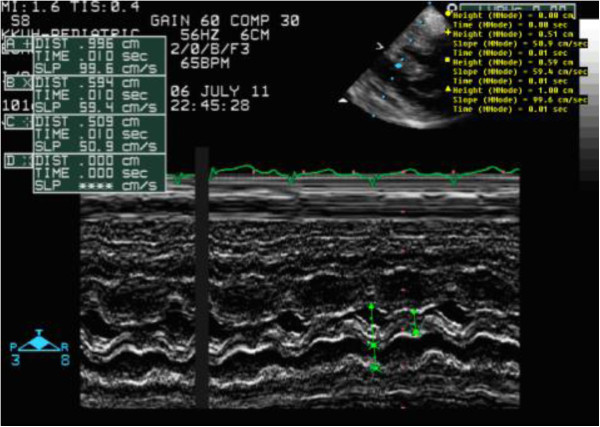
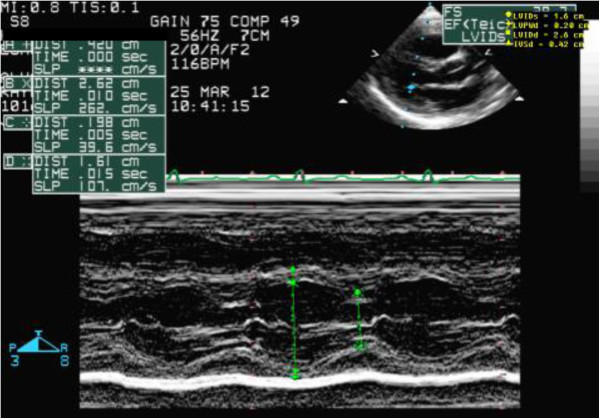
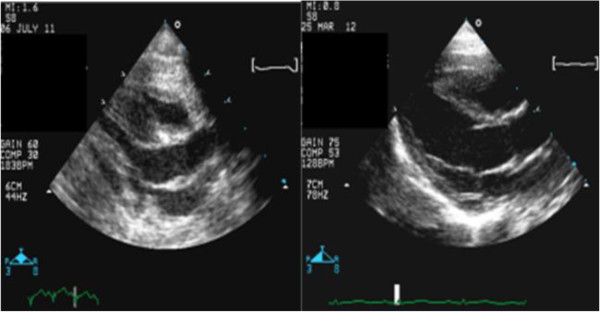
Case presentation: We report two Saudi siblings with TT1. Case 1 was a male infant who presented at 2 months old with fever, vomiting and refusal of feeding. Examination revealed a sick-looking infant with signs of severe dehydration and hypovolemic shock. He was jaundiced, and had hepatomegaly and elevated liver enzymes. Echocardiography was performed in light of a lack of response to inotropes, and revealed biventricular and interventricular septal hypertrophies. The ventricular ejection fraction was 65%. Urine organic acid analysis showed elevated succinylacetone, consistent with a diagnosis of TT1. An FAH gene study identified a c.1 A > G homozygous mutation. This patient responded well to intensive cardiorespiratory therapy, tyrosine-free formula, and oral 2-nitro-4- trifluoromethylbenzyl 1, 3 cyclohexanedione (NTBC). Echocardiographic findings reverted to normal after 4 weeks. Case 2 was the younger brother of Case 1, and was born 6 months after his brother had been confirmed with tyrosinemia. Pregnancy and delivery were uneventful. Serum amino acid and organic acid analyses 4 days after birth confirmed tyrosinemia. DNA analysis identified a c.1 A > G homozygous mutation, as in his brother. Echocardiography was normal. Special formula and NTBC were commenced on day 7 of life. The infant remained asymptomatic after 9 months of follow-up.
Conclusions: These cases highlight TT1 as a treatable cause of cardiomyopathy in children. It also supports the idea that early diagnosis and treatment may prevent the development of cardiomyopathy associated with tyrosinemia.
Figures
References
-
- Mitchell GA, Grompe M, Lambert M, Tanguay RM. In: Hypertyrosinemia : the metabolic and molecular bases of inherited disease. 5. Scriver CR, Beaudet A, Sly WS, Valle D, editor. New York: McGraw Hill; 2001. pp. 1777–1806.
-
- Nugent AW, Daubeney PEF, Chondros P, Carlin JB, Cheung M, Wilkinson LC, Davis AM, Kahler SG, Chow CW, Wilkinson JL, Weintraub RG. National Australian childhood cardiomyopathy study.: the epidemiology of childhood cardiomyopathy in Australia. N Engl J Med. 2003;348(17):1639–1646. doi: 10.1056/NEJMoa021737. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
