

Mapping yeast transcriptional networks
- PMID: 24018767
- PMCID: PMC3761317
- DOI: 10.1534/genetics.113.153262
Mapping yeast transcriptional networks
Abstract
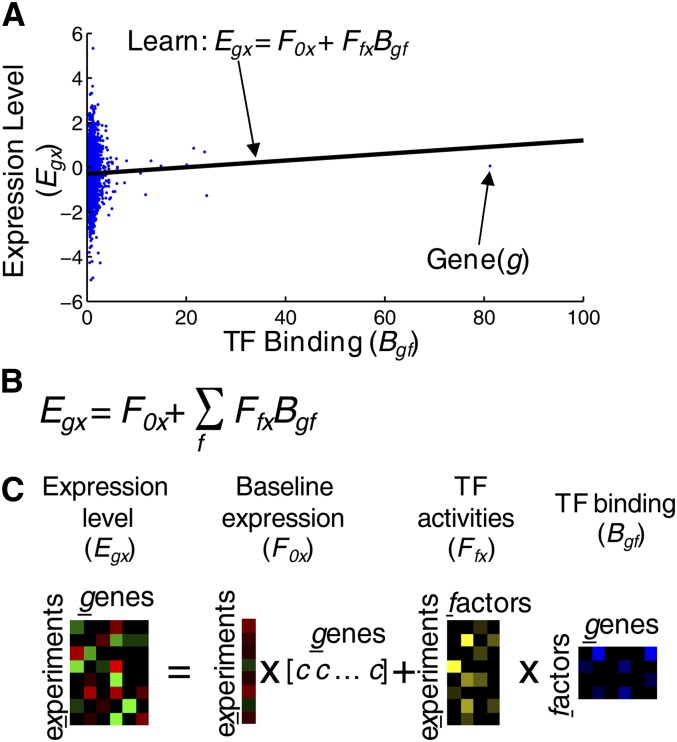
The term "transcriptional network" refers to the mechanism(s) that underlies coordinated expression of genes, typically involving transcription factors (TFs) binding to the promoters of multiple genes, and individual genes controlled by multiple TFs. A multitude of studies in the last two decades have aimed to map and characterize transcriptional networks in the yeast Saccharomyces cerevisiae. We review the methodologies and accomplishments of these studies, as well as challenges we now face. For most yeast TFs, data have been collected on their sequence preferences, in vivo promoter occupancy, and gene expression profiles in deletion mutants. These systematic studies have led to the identification of new regulators of numerous cellular functions and shed light on the overall organization of yeast gene regulation. However, many yeast TFs appear to be inactive under standard laboratory growth conditions, and many of the available data were collected using techniques that have since been improved. Perhaps as a consequence, comprehensive and accurate mapping among TF sequence preferences, promoter binding, and gene expression remains an open challenge. We propose that the time is ripe for renewed systematic efforts toward a complete mapping of yeast transcriptional regulatory mechanisms.
Keywords: chromatin; gene expression; regulatory networks; transcription factors; yeast.
Figures
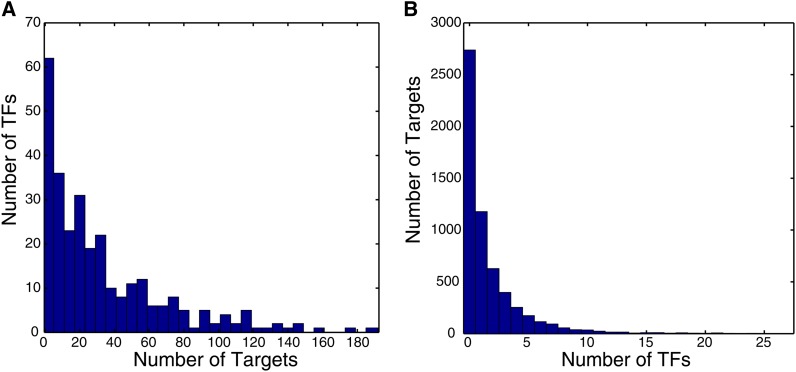
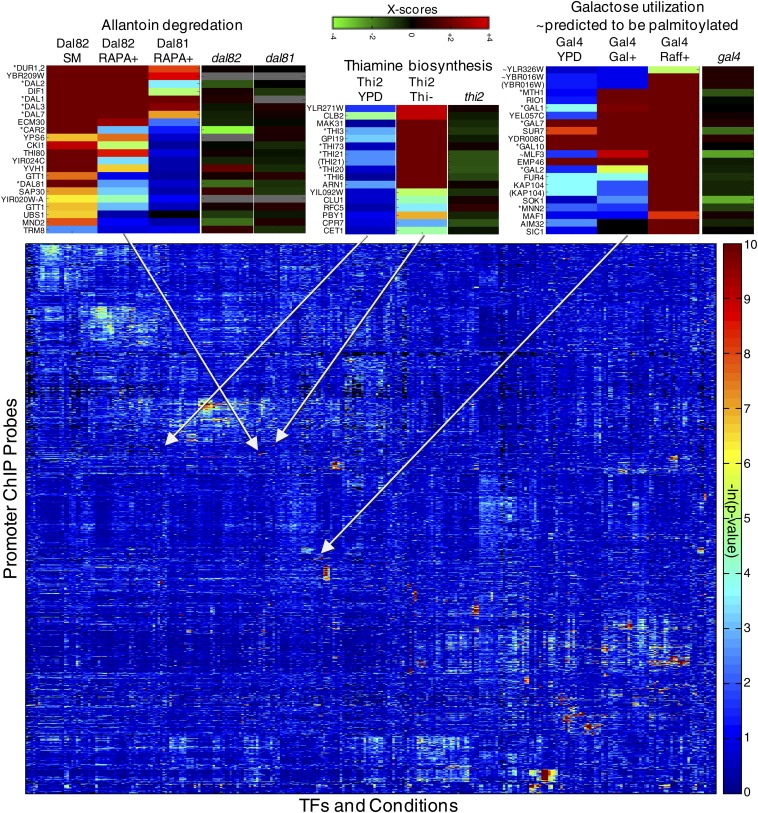
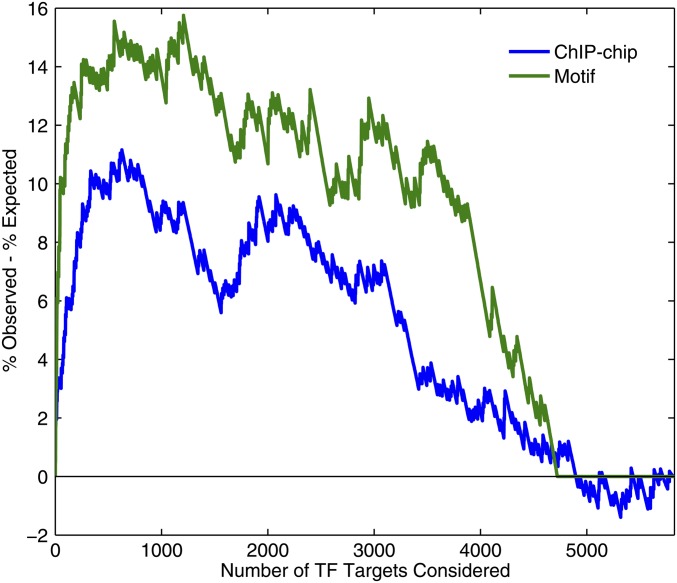
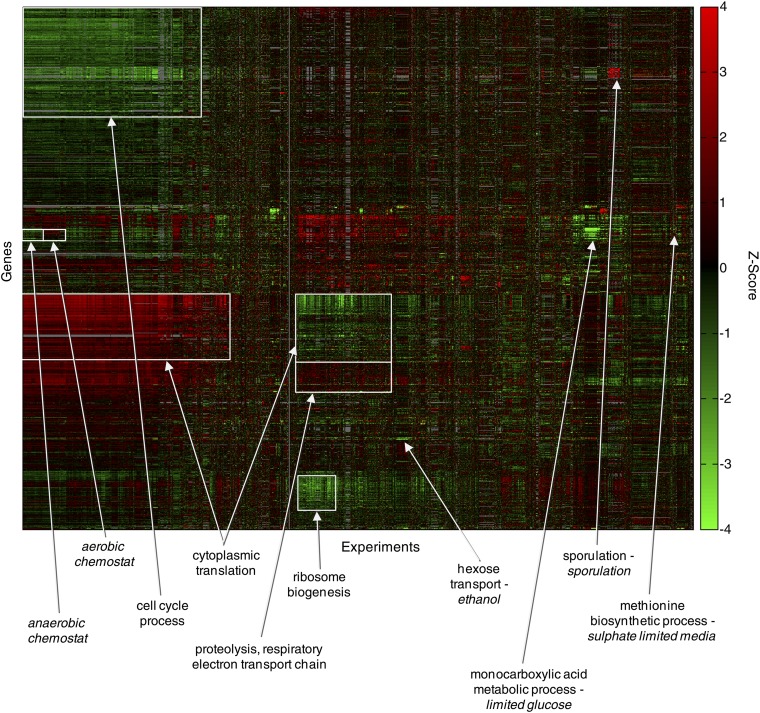
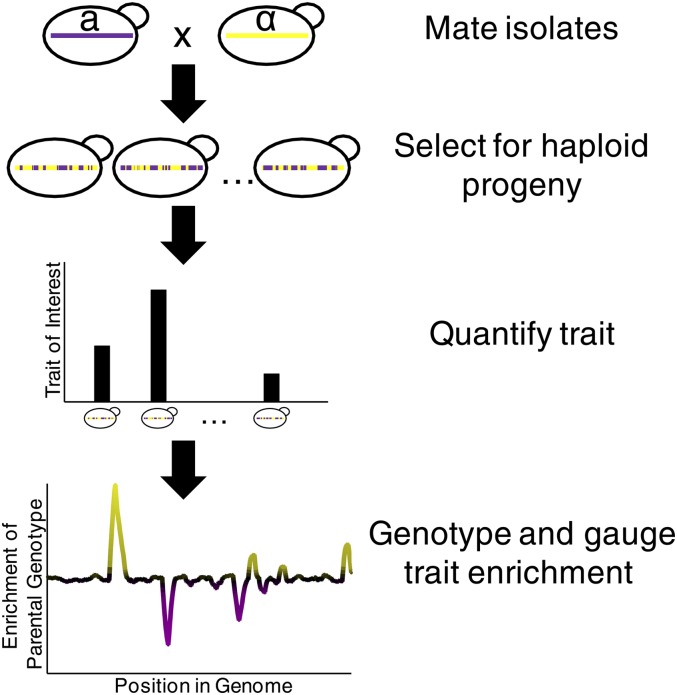
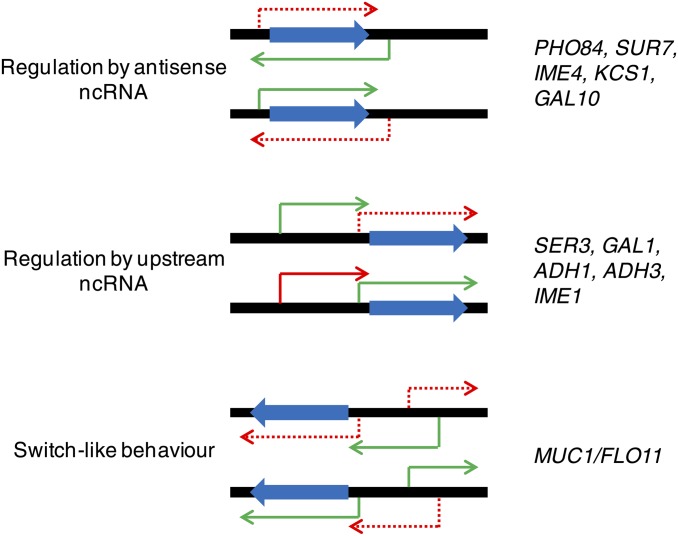
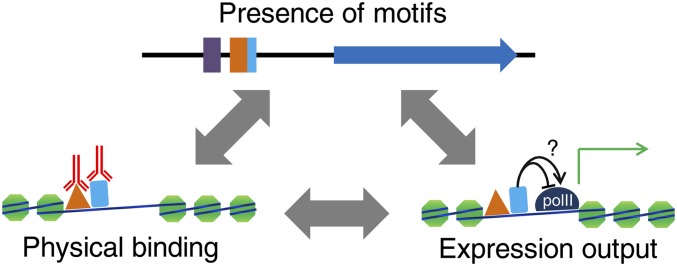
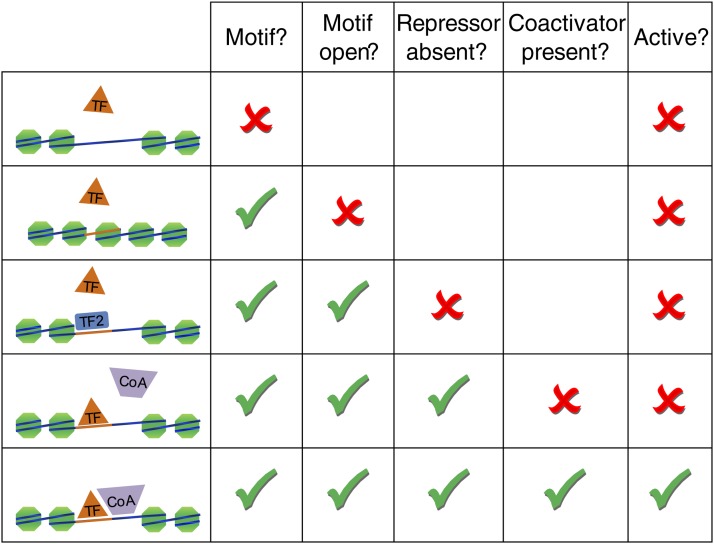
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
