

Association between early-onset Parkinson disease and 22q11.2 deletion syndrome: identification of a novel genetic form of Parkinson disease and its clinical implications
- PMID: 24018986
- PMCID: PMC4464823
- DOI: 10.1001/jamaneurol.2013.3646
Association between early-onset Parkinson disease and 22q11.2 deletion syndrome: identification of a novel genetic form of Parkinson disease and its clinical implications
Abstract
Importance: Clinical case reports of parkinsonism co-occurring with hemizygous 22q11.2 deletions and the associated multisystem syndrome, 22q11.2 deletion syndrome (22q11.2DS), suggest that 22q11.2 deletions may lead to increased risk of early-onset Parkinson disease (PD). The frequency of PD and its neuropathological presentation remain unknown in this common genetic condition.
Objective: To evaluate a possible association between 22q11.2 deletions and PD.
Design, setting, and participants: An observational study of the occurrence of PD in the world's largest cohort of well-characterized adults with a molecularly confirmed diagnosis of 22q11.2DS (n = 159 [6 with postmortem tissue]; age range, 18.1-68.6 years) was conducted in Toronto, Ontario, Canada. Rare postmortem brain tissue from individuals with 22q11.2DS and a clinical history of PD was investigated for neurodegenerative changes and compared with that from individuals with no history of a movement disorder.
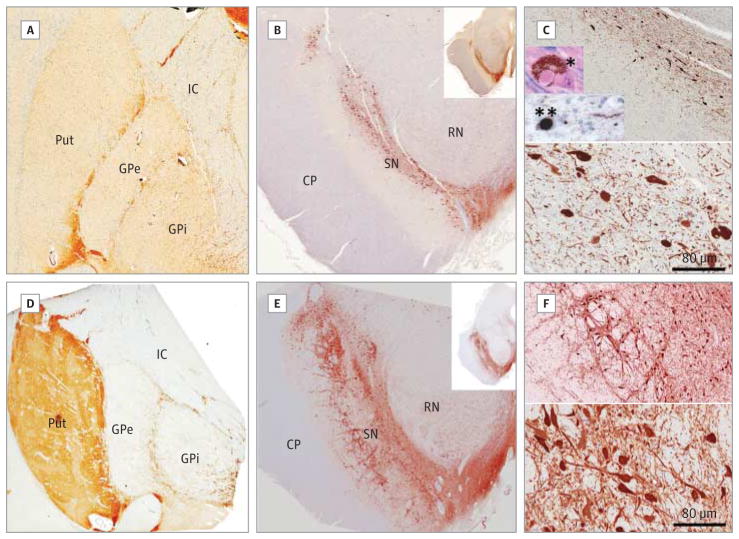
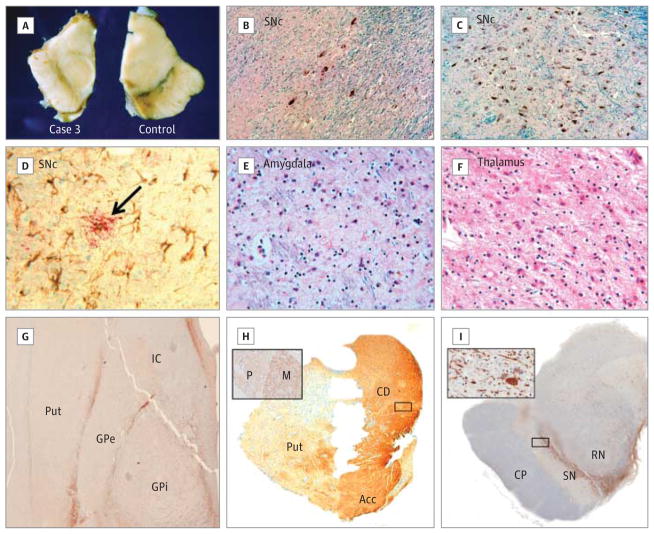
Main outcomes and measures: A clinical diagnosis of PD made by a neurologist and neuropathological features of PD. RESULTS Adults with 22q11.2DS had a significantly elevated occurrence of PD compared with standard population estimates (standardized morbidity ratio = 69.7; 95% CI, 19.0-178.5). All cases showed early onset and typical PD symptom pattern, treatment response, and course. All were negative for family history of PD and known pathogenic PD-related mutations. The common use of antipsychotics in patients with 22q11.2DS to manage associated psychiatric symptoms delayed diagnosis of PD by up to 10 years. Postmortem brain tissue revealed classic loss of midbrain dopaminergic neurons in all 3 postmortem 22q11.2DS-PD cases. Typical α-synuclein-positive Lewy bodies were present in the expected distribution in 2 cases but absent in another.
Conclusions and relevance: These findings suggest that 22q11.2 deletions represent a novel genetic risk factor for early-onset PD with variable neuropathological presentation reminiscent of LRRK2-associated PD neuropathology. Individuals with early-onset PD and classic features of 22q11.2DS should be considered for genetic testing, and those with a known 22q11.2 deletion should be monitored for the development of parkinsonian symptoms. Molecular studies of the implicated genes, including DGCR8, may help shed light on the underlying pathophysiology of PD in 22q11.2DS and idiopathic PD.
Conflict of interest statement
Figures
Comment in
-
Structural variation and the expanding genomic architecture of Parkinson disease.JAMA Neurol. 2013 Nov;70(11):1355-6. doi: 10.1001/jamaneurol.2013.4263. JAMA Neurol. 2013. PMID: 24018918 No abstract available.
References
-
- Lai BC, Schulzer M, Marion S, Teschke K, Tsui JK. The prevalence of Parkinson’s disease in British Columbia, Canada, estimated by using drug tracer methodology. Parkinsonism Relat Disord. 2003;9(4):233–238. - PubMed
-
- Wirdefeldt K, Adami HO, Cole P, Trichopoulos D, Mandel J. Epidemiology and etiology of Parkinson’s disease: a review of the evidence. Eur J Epidemiol. 2011;26(suppl 1):S1–S58. - PubMed
-
- Macedo MG, Verbaan D, Fang Y, et al. Genotypic and phenotypic characteristics of Dutch patients with early onset Parkinson’s disease. Mov Disord. 2009;24(2):196–203. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
