

Mechanism for activation of mutated epidermal growth factor receptors in lung cancer
- PMID: 24019492
- PMCID: PMC3780914
- DOI: 10.1073/pnas.1220050110
Mechanism for activation of mutated epidermal growth factor receptors in lung cancer
Erratum in
- Proc Natl Acad Sci U S A. 2013 Dec 10;110(50):20344
Abstract
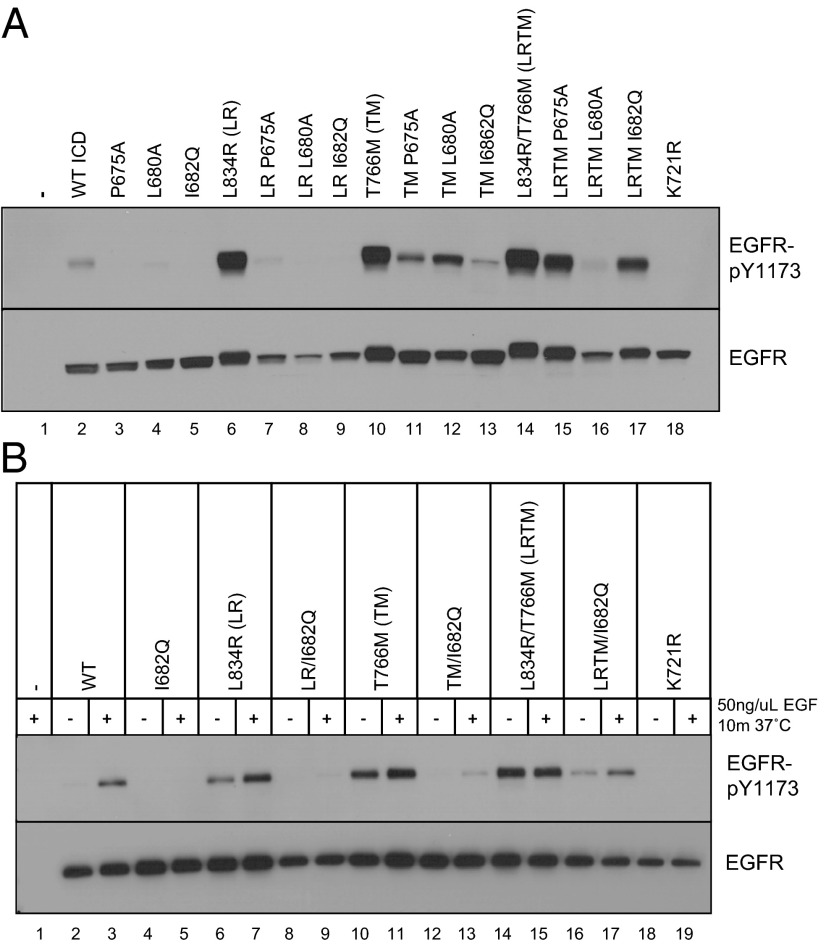
The initiation of epidermal growth factor receptor (EGFR) kinase activity proceeds via an asymmetric dimerization mechanism in which a "donor" tyrosine kinase domain (TKD) contacts an "acceptor" TKD, leading to its activation. In the context of a ligand-induced dimer, identical wild-type EGFR TKDs are thought to assume the donor or acceptor roles in a random manner. Here, we present biochemical reconstitution data demonstrating that activated EGFR mutants found in lung cancer preferentially assume the acceptor role when coexpressed with WT EGFR. Mutated EGFRs show enhanced association with WT EGFR, leading to hyperphosphorylation of the WT counterpart. Mutated EGFRs also hyperphosphorylate the related erythroblastic leukemia viral oncogene (ErbB) family member, ErbB-2, in a similar manner. This directional "superacceptor activity" is particularly pronounced in the drug-resistant L834R/T766M mutant. A 4-Å crystal structure of this mutant in the active conformation reveals an asymmetric dimer interface that is essentially the same as that in WT EGFR. Asymmetric dimer formation induces an allosteric conformational change in the acceptor subunit. Thus, superacceptor activity likely arises simply from a lower energetic cost associated with this conformational change in the mutant EGFR compared with WT, rather than from any structural alteration that impairs the donor role of the mutant. Collectively, these findings define a previously unrecognized mode of mutant-specific intermolecular regulation for ErbB receptors, knowledge of which could potentially be exploited for therapeutic benefit.
Keywords: TKI; WZ-4002; lapatinib; mutation.
Conflict of interest statement
Conflict of interest statement: Rights to testing of EGFR T790M mutations were licensed by Memorial Sloan-Kettering Cancer Center on behalf of W.P. and others to MolecularMD.
Figures






Comment in
-
EGFR lung cancer mutants get specialized.Proc Natl Acad Sci U S A. 2013 Sep 17;110(38):15169-70. doi: 10.1073/pnas.1314719110. Epub 2013 Sep 10. Proc Natl Acad Sci U S A. 2013. PMID: 24023066 Free PMC article. No abstract available.
References
-
- Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer. 2007;7(3):169–181. - PubMed
-
- Mitsudomi T, et al. West Japan Oncology Group Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): An open label, randomised phase 3 trial. Lancet Oncol. 2010;11(2):121–128. - PubMed
-
- Mok TS, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361(10):947–957. - PubMed
-
- Rosell R, Viteri S, Molina MA, Benlloch S, Taron M. Epidermal growth factor receptor tyrosine kinase inhibitors as first-line treatment in advanced nonsmall-cell lung cancer. Curr Opin Oncol. 2010;22(2):112–120. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
- R01 CA079992/CA/NCI NIH HHS/United States
- U54 CA143798/CA/NCI NIH HHS/United States
- P01 CA154303/CA/NCI NIH HHS/United States
- P30-CA68485/CA/NCI NIH HHS/United States
- R01 CA121210/CA/NCI NIH HHS/United States
- P50 CA090949/CA/NCI NIH HHS/United States
- R01 CA116020/CA/NCI NIH HHS/United States
- P30 CA068485/CA/NCI NIH HHS/United States
- R01-CA116020/CA/NCI NIH HHS/United States
- U54-CA143798/CA/NCI NIH HHS/United States
- R01-CA121210/CA/NCI NIH HHS/United States
- CA90949/CA/NCI NIH HHS/United States
- P01-CA129243/CA/NCI NIH HHS/United States
- P01-CA154303/CA/NCI NIH HHS/United States
- P01 CA129243/CA/NCI NIH HHS/United States
- R01-CA079992/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
