

The epidemiology of premature aging and associated comorbidities
- PMID: 24019745
- PMCID: PMC3760297
- DOI: 10.2147/CIA.S37213
The epidemiology of premature aging and associated comorbidities
Abstract
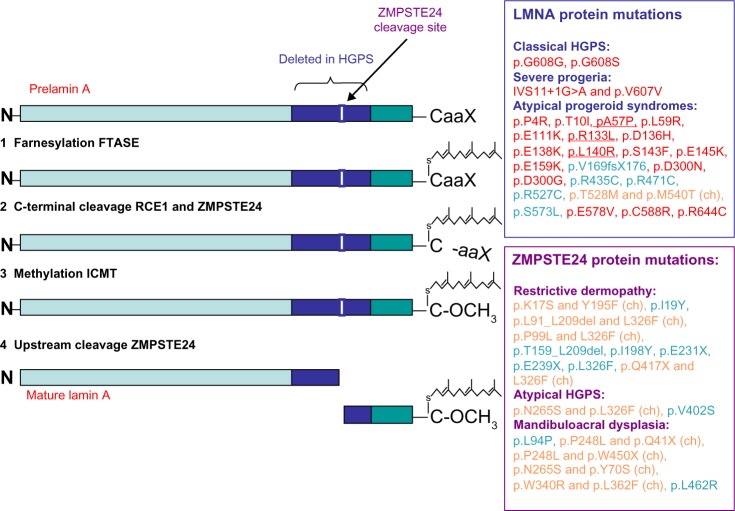
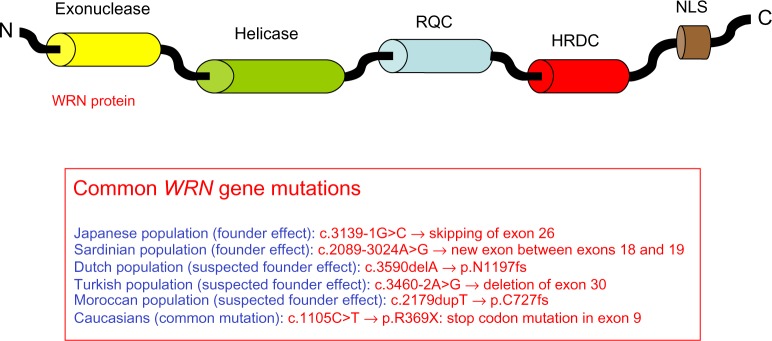
Hutchinson-Gilford Progeria Syndrome and Werner syndrome, also known as childhood- and adulthood-progeria, respectively, represent two of the best characterized human progeroid diseases with clinical features mimicking physiological aging at an early age. The discovery of their genetic basis has led to the identification of several gene mutations leading to a spectrum of progeroid phenotypes ranging from moderate and mild-severe to very aggressive forms. In parallel, the creation of disease registers and databases provided available data for the design of relatively large-scale epidemiological studies, thereby allowing a better understanding of the nature and frequency of the premature aging-associated signs and symptoms. The aim of this article is to review the most recent findings concerning the epidemiology of premature aging disorders, their genetic basis, and the most recent reports on the frequency of associated diseases.
Keywords: Hutchinson–Gilford Progeria Syndrome; Werner syndrome; atherosclerosis; cancer; cardiovascular diseases; epidemiology; genetics; premature aging disorders; sign and symptoms.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
