

Comparing methods for estimating R0 from the size distribution of subcritical transmission chains
- PMID: 24021520
- PMCID: PMC3821076
- DOI: 10.1016/j.epidem.2013.05.002
Comparing methods for estimating R0 from the size distribution of subcritical transmission chains
Abstract
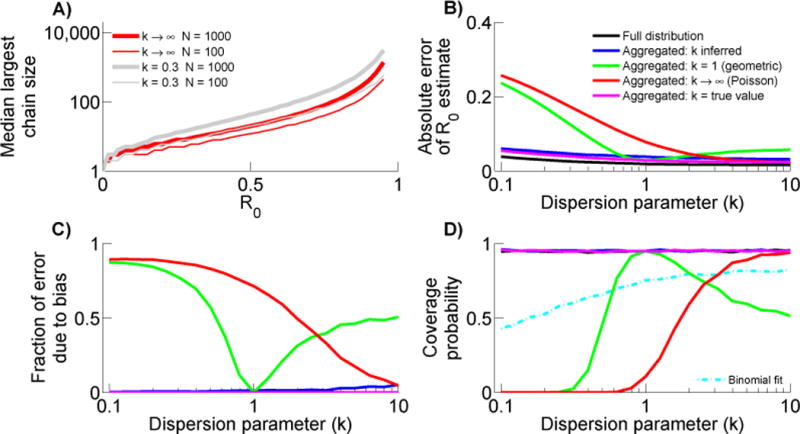
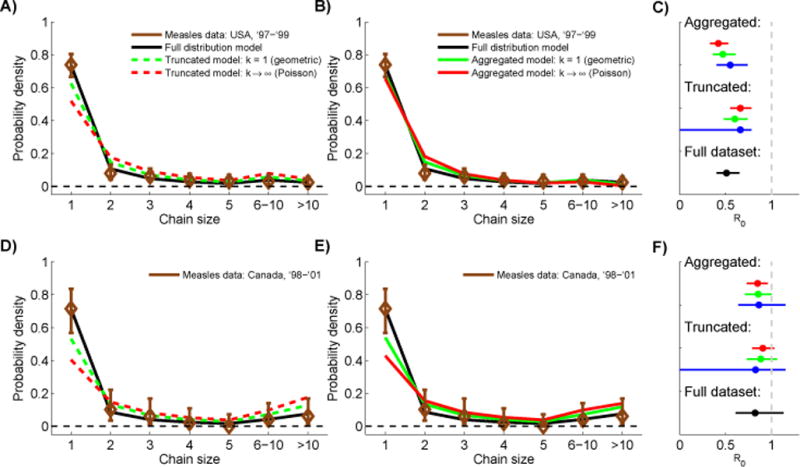
Many diseases exhibit subcritical transmission (i.e. 0<R0<1) so that infections occur as self-limited 'stuttering chains'. Given an ensemble of stuttering chains, information about the number of cases in each chain can be used to infer R0, which is of crucial importance for monitoring the risk that a disease will emerge to establish endemic circulation. However, the challenge of imperfect case detection has led authors to adopt a variety of work-around measures when inferring R0, such as discarding data on isolated cases or aggregating intermediate-sized chains together. Each of these methods has the potential to introduce bias, but a quantitative comparison of these approaches has not been reported. By adapting a model based on a negative binomial offspring distribution that permits a variable degree of transmission heterogeneity, we present a unified analysis of existing R0 estimation methods. Simulation studies show that the degree of transmission heterogeneity, when improperly modeled, can significantly impact the bias of R0 estimation methods designed for imperfect observation. These studies also highlight the importance of isolated cases in assessing whether an estimation technique is consistent with observed data. Analysis of data from measles outbreaks shows that likelihood scores are highest for models that allow a flexible degree of transmission heterogeneity. Aggregating intermediate sized chains often has similar performance to analyzing a complete chain size distribution. However, truncating isolated cases is beneficial only when surveillance systems clearly favor full observation of large chains but not small chains. Meanwhile, if data on the type and proportion of cases that are unobserved were known, we demonstrate that maximum likelihood inference of R0 could be adjusted accordingly. This motivates the need for future empirical and theoretical work to quantify observation error and incorporate relevant mechanisms into stuttering chain models used to estimate transmission parameters.
Keywords: Basic reproductive number; Imperfect observation; Measles; Stuttering chain; Transmission heterogeneity.
Copyright © 2013 Elsevier B.V. All rights reserved.
Figures







References
-
- Ball FG, Britton T, O’Neill PD. Empty confidence sets for epidemics, branching processes and brownian motion. Biometrika. 2002;89:211–224.
-
- Becker N. On parametric estimation for mortal branching processes. Biometrika. 1974;61:393–399. http://biomet.oxfordjournals.org/content/61/2/393.full.pdf+html.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
