

Oxidized Ca(2+)/calmodulin-dependent protein kinase II triggers atrial fibrillation
- PMID: 24030498
- PMCID: PMC3876034
- DOI: 10.1161/CIRCULATIONAHA.113.003313
Oxidized Ca(2+)/calmodulin-dependent protein kinase II triggers atrial fibrillation
Abstract
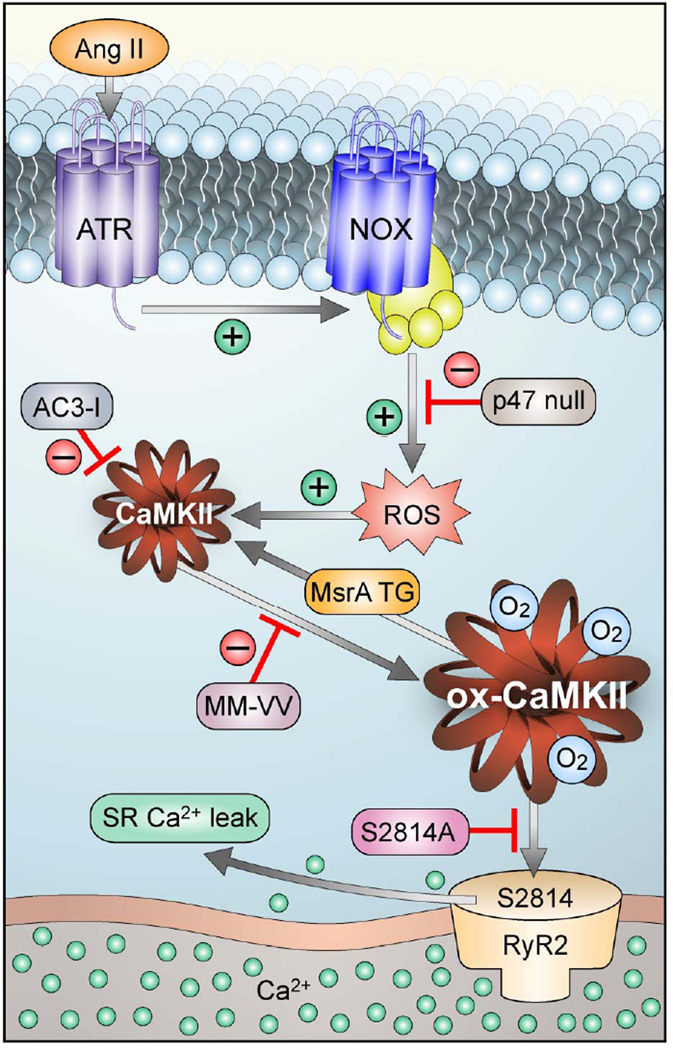
Background: Atrial fibrillation (AF) is a growing public health problem without adequate therapies. Angiotensin II and reactive oxygen species are validated risk factors for AF in patients, but the molecular pathways connecting reactive oxygen species and AF are unknown. The Ca(2+)/calmodulin-dependent protein kinase II (CaMKII) has recently emerged as a reactive oxygen species-activated proarrhythmic signal, so we hypothesized that oxidized CaMKIIδ could contribute to AF.
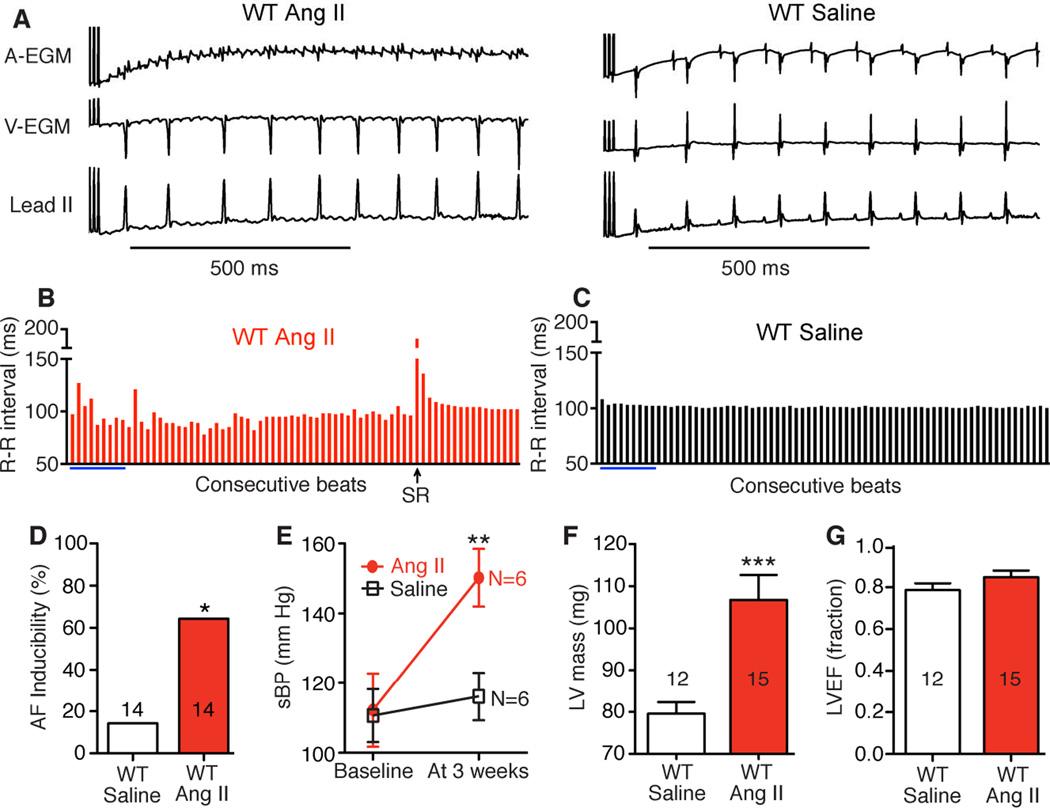
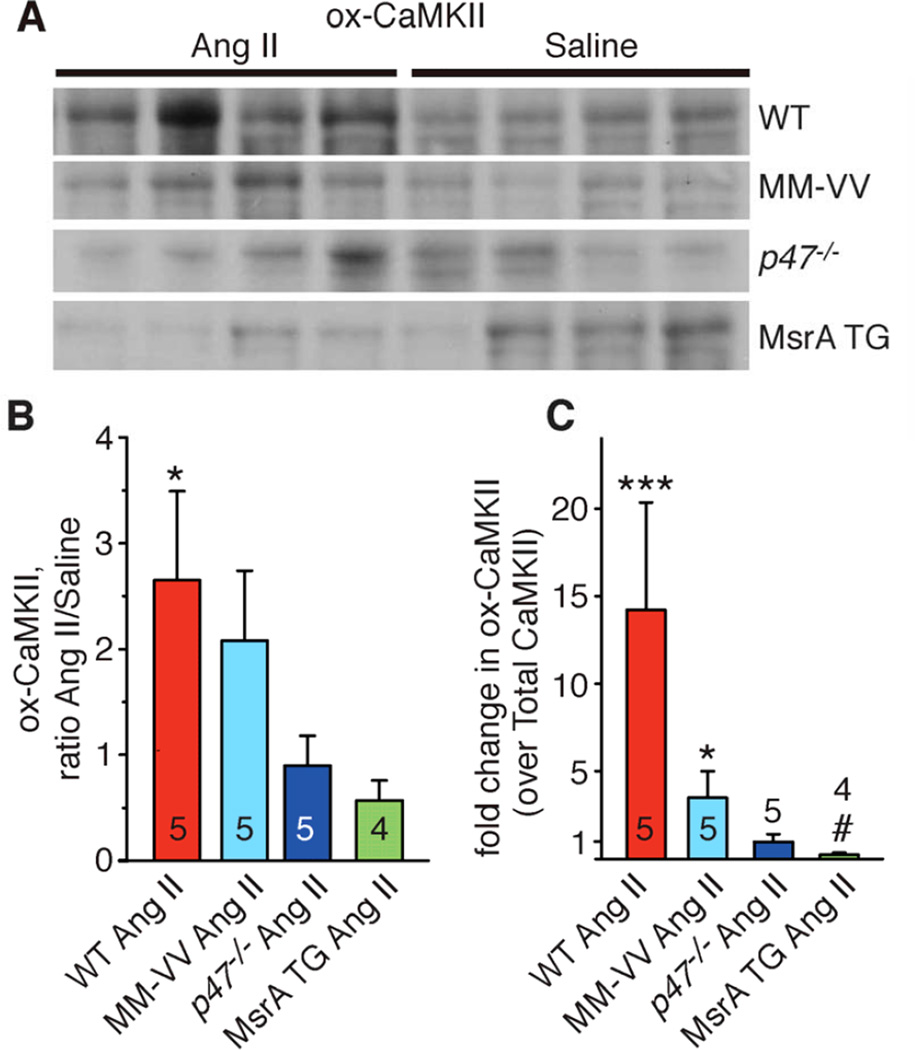
Methods and results: We found that oxidized CaMKII was increased in atria from AF patients compared with patients in sinus rhythm and from mice infused with angiotensin II compared with mice infused with saline. Angiotensin II-treated mice had increased susceptibility to AF compared with saline-treated wild-type mice, establishing angiotensin II as a risk factor for AF in mice. Knock-in mice lacking critical oxidation sites in CaMKIIδ (MM-VV) and mice with myocardium-restricted transgenic overexpression of methionine sulfoxide reductase A, an enzyme that reduces oxidized CaMKII, were resistant to AF induction after angiotensin II infusion.
Conclusions: Our studies suggest that CaMKII is a molecular signal that couples increased reactive oxygen species with AF and that therapeutic strategies to decrease oxidized CaMKII may prevent or reduce AF.
Keywords: angiotensin II; arrhythmias, cardiac; atrial fibrillation; calcium-calmodulin-dependent protein kinase type II; reactive oxygen species.
Conflict of interest statement
Figures




Comment in
-
Oxidative stress and atrial fibrillation: finding a missing piece to the puzzle.Circulation. 2013 Oct 15;128(16):1724-6. doi: 10.1161/CIRCULATIONAHA.113.005837. Epub 2013 Sep 12. Circulation. 2013. PMID: 24030497 Free PMC article. No abstract available.
References
-
- Benjamin EJ, Wolf PA, D'Agostino RB, Silbershatz H, Kannel WB, Levy D. Impact of atrial fibrillation on the risk of death: the Framingham Heart Study. Circulation. 1998;98:946–952. - PubMed
-
- Khatib R, Joseph P, Briel M, Yusuf S, Healey J. Blockade of the renin-angiotensin-aldosterone system (RAAS) for primary prevention of non-valvular atrial fibrillation: A systematic review and meta analysis of randomized controlled trials. Int J Cardiol. 2013;165:17–24. - PubMed
-
- Nattel S. New ideas about atrial fibrillation 50 years on. Nature. 2002;415:219–226. - PubMed
-
- Shimano M, Shibata R, Inden Y, Yoshida N, Uchikawa T, Tsuji Y, Murohara T. Reactive oxidative metabolites are associated with atrial conduction disturbance in patients with atrial fibrillation. Heart Rhythm. 2009;6:935–940. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 HL079031/HL/NHLBI NIH HHS/United States
- R01 HL096652/HL/NHLBI NIH HHS/United States
- R01-HL070250/HL/NHLBI NIH HHS/United States
- R01 HL113001/HL/NHLBI NIH HHS/United States
- RR026293/RR/NCRR NIH HHS/United States
- R01 HL091947/HL/NHLBI NIH HHS/United States
- R01 HL070250/HL/NHLBI NIH HHS/United States
- R01-HL0909050/HL/NHLBI NIH HHS/United States
- R01 HL089598/HL/NHLBI NIH HHS/United States
- R01-HL096652/HL/NHLBI NIH HHS/United States
- R01-HL071140/HL/NHLBI NIH HHS/United States
- S10 RR026293/RR/NCRR NIH HHS/United States
- R01 HL071140/HL/NHLBI NIH HHS/United States
- R01-HL091947/HL/NHLBI NIH HHS/United States
- R01-HL 079031/HL/NHLBI NIH HHS/United States
- R01-HL089598/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
