

Next generation sequencing for molecular diagnosis of neurological disorders using ataxias as a model
- PMID: 24030952
- PMCID: PMC3784284
- DOI: 10.1093/brain/awt236
Next generation sequencing for molecular diagnosis of neurological disorders using ataxias as a model
Abstract
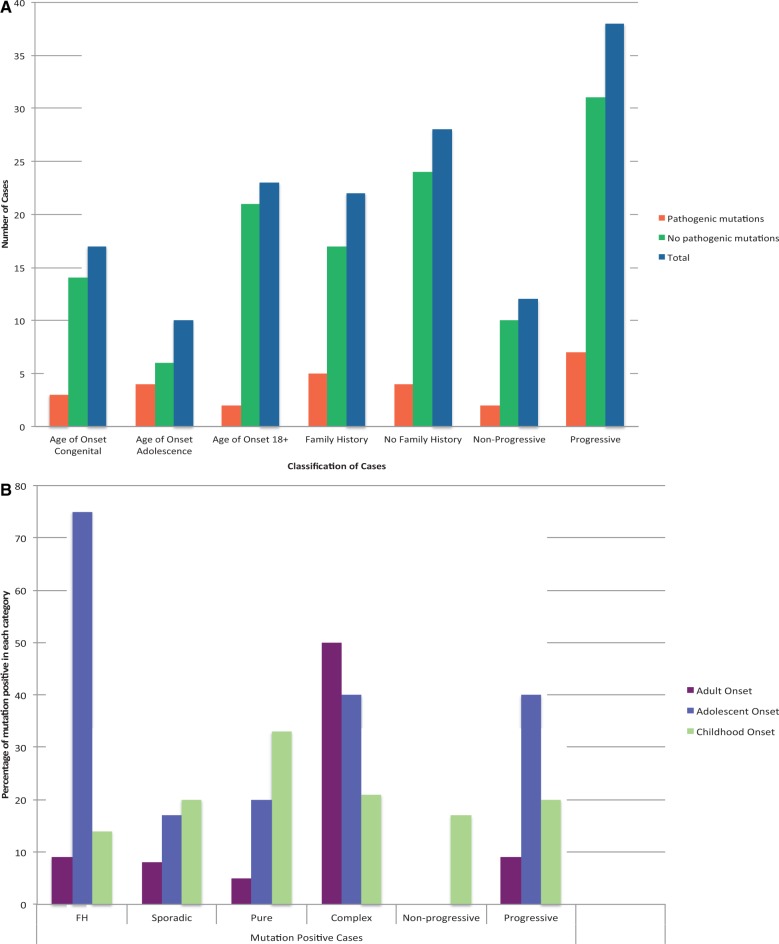
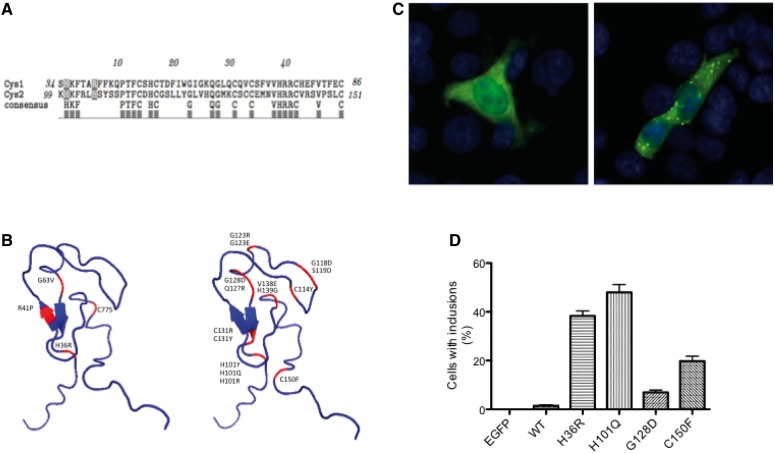
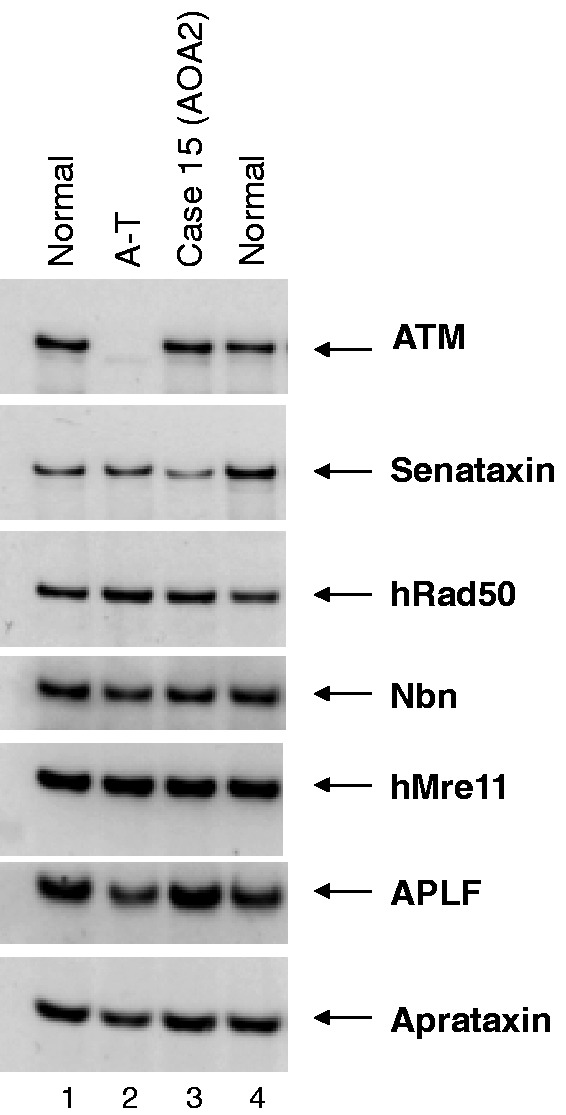
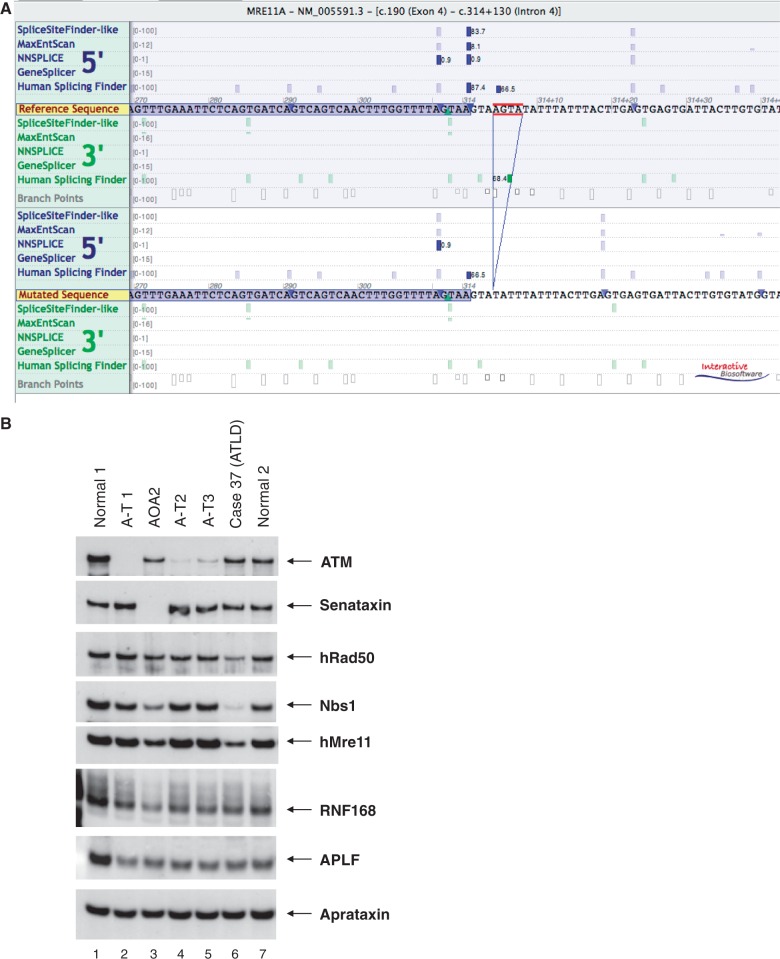
Many neurological conditions are caused by immensely heterogeneous gene mutations. The diagnostic process is often long and complex with most patients undergoing multiple invasive and costly investigations without ever reaching a conclusive molecular diagnosis. The advent of massively parallel, next-generation sequencing promises to revolutionize genetic testing and shorten the 'diagnostic odyssey' for many of these patients. We performed a pilot study using heterogeneous ataxias as a model neurogenetic disorder to assess the introduction of next-generation sequencing into clinical practice. We captured 58 known human ataxia genes followed by Illumina Next-Generation Sequencing in 50 highly heterogeneous patients with ataxia who had been extensively investigated and were refractory to diagnosis. All cases had been tested for spinocerebellar ataxia 1-3, 6, 7 and Friedrich's ataxia and had multiple other biochemical, genetic and invasive tests. In those cases where we identified the genetic mutation, we determined the time to diagnosis. Pathogenicity was assessed using a bioinformatics pipeline and novel variants were validated using functional experiments. The overall detection rate in our heterogeneous cohort was 18% and varied from 8.3% in those with an adult onset progressive disorder to 40% in those with a childhood or adolescent onset progressive disorder. The highest detection rate was in those with an adolescent onset and a family history (75%). The majority of cases with detectable mutations had a childhood onset but most are now adults, reflecting the long delay in diagnosis. The delays were primarily related to lack of easily available clinical testing, but other factors included the presence of atypical phenotypes and the use of indirect testing. In the cases where we made an eventual diagnosis, the delay was 3-35 years (mean 18.1 years). Alignment and coverage metrics indicated that the capture and sequencing was highly efficient and the consumable cost was ∼£400 (€460 or US$620). Our pathogenicity interpretation pathway predicted 13 different mutations in eight different genes: PRKCG, TTBK2, SETX, SPTBN2, SACS, MRE11, KCNC3 and DARS2 of which nine were novel including one causing a newly described recessive ataxia syndrome. Genetic testing using targeted capture followed by next-generation sequencing was efficient, cost-effective, and enabled a molecular diagnosis in many refractory cases. A specific challenge of next-generation sequencing data is pathogenicity interpretation, but functional analysis confirmed the pathogenicity of novel variants showing that the pipeline was robust. Our results have broad implications for clinical neurology practice and the approach to diagnostic testing.
Keywords: ataxia; autosomal dominant cerebellar ataxia; autosomal recessive cerebellar ataxia; diagnosis; genetics.
Figures
Comment in
-
Genetics: Utility of next-generation sequencing in ataxias.Nat Rev Neurol. 2013 Nov;9(11):614-5. doi: 10.1038/nrneurol.2013.212. Epub 2013 Oct 15. Nat Rev Neurol. 2013. PMID: 24126630 No abstract available.
References
-
- Alonso I, Costa C, Gomes A, Ferro A, Seixas AI, Silva S, et al. A novel H101Q mutation causes PKCgamma loss in spinocerebellar ataxia type 14. J Hum Genet. 2005;50:523–9. - PubMed
-
- Anheim M, Fleury M, Monga B, Laugel V, Chaigne D, Rodier G, et al. Epidemiological, clinical, paraclinical and molecular study of a cohort of 102 patients affected with autosomal recessive progressive cerebellar ataxia from Alsace, Eastern France: implications for clinical management. Neurogenetics. 2010;11:1–12. - PubMed
-
- Antonellis A, Green ED. The role of aminoacyl-tRNA synthetases in genetic diseases. Annu Rev Genomics Hum Genet. 2008;9:87–107. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
