

A systematic approach to assessing the clinical significance of genetic variants
- PMID: 24033266
- PMCID: PMC3995020
- DOI: 10.1111/cge.12257
A systematic approach to assessing the clinical significance of genetic variants
Abstract
Molecular genetic testing informs diagnosis, prognosis, and risk assessment for patients and their family members. Recent advances in low-cost, high-throughput DNA sequencing and computing technologies have enabled the rapid expansion of genetic test content, resulting in dramatically increased numbers of DNA variants identified per test. To address this challenge, our laboratory has developed a systematic approach to thorough and efficient assessments of variants for pathogenicity determination. We first search for existing data in publications and databases including internal, collaborative and public resources. We then perform full evidence-based assessments through statistical analyses of observations in the general population and disease cohorts, evaluation of experimental data from in vivo or in vitro studies, and computational predictions of potential impacts of each variant. Finally, we weigh all evidence to reach an overall conclusion on the potential for each variant to be disease causing. In this report, we highlight the principles of variant assessment, address the caveats and pitfalls, and provide examples to illustrate the process. By sharing our experience and providing a framework for variant assessment, including access to a freely available customizable tool, we hope to help move towards standardized and consistent approaches to variant assessment.
Keywords: (4−9) clinical interpretation; gain-of-function (GOF); genetic variant; loss of function (LOF); next-generation sequencing (NGS); sequence analysis; variant assessment; variant of uncertain significance (VUS).
© 2013 John Wiley & Sons A/S. Published by John Wiley & Sons Ltd.
Conflict of interest statement
HD, JS, HM, MAK, TJP, BHF, HLR and MSL are employed by fee-for-service laboratories performing clinical sequencing services. Several individuals serve on advisory boards or in other capacities for companies providing sequencing or other genetic services (HLR – BioBase, Clinical Future, Complete Genomics, GenomeQuest, Illumina, Ingenuity, Knome, Omicia; BF – InVitae; JS – LabCorp).
Figures
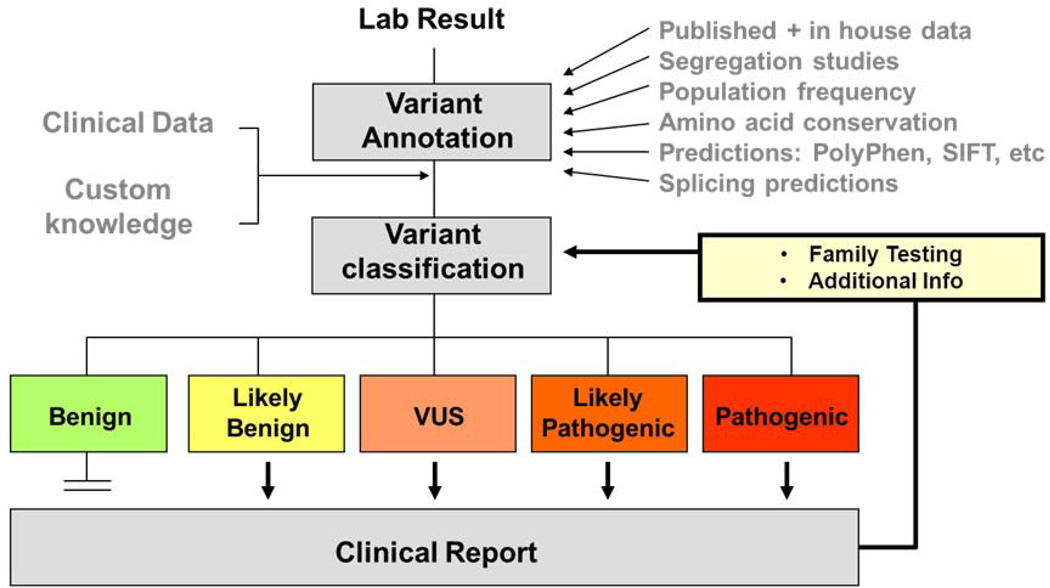
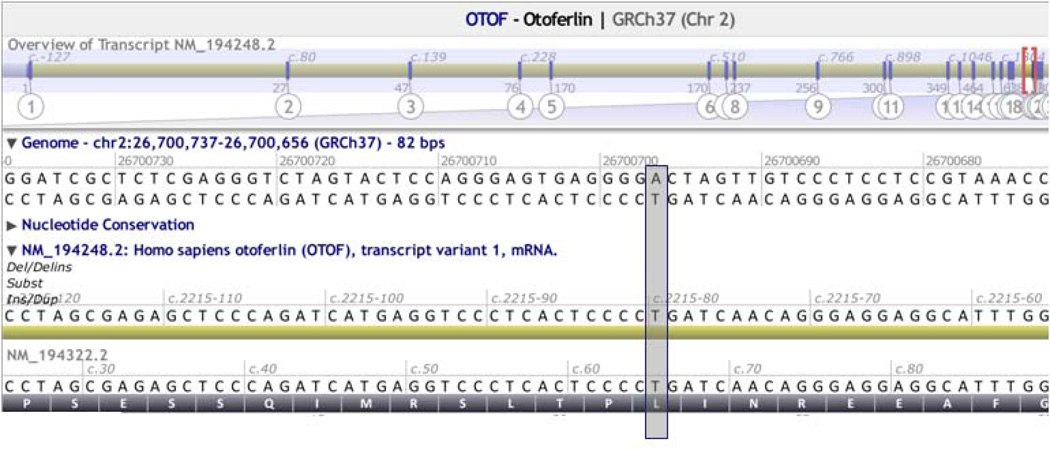
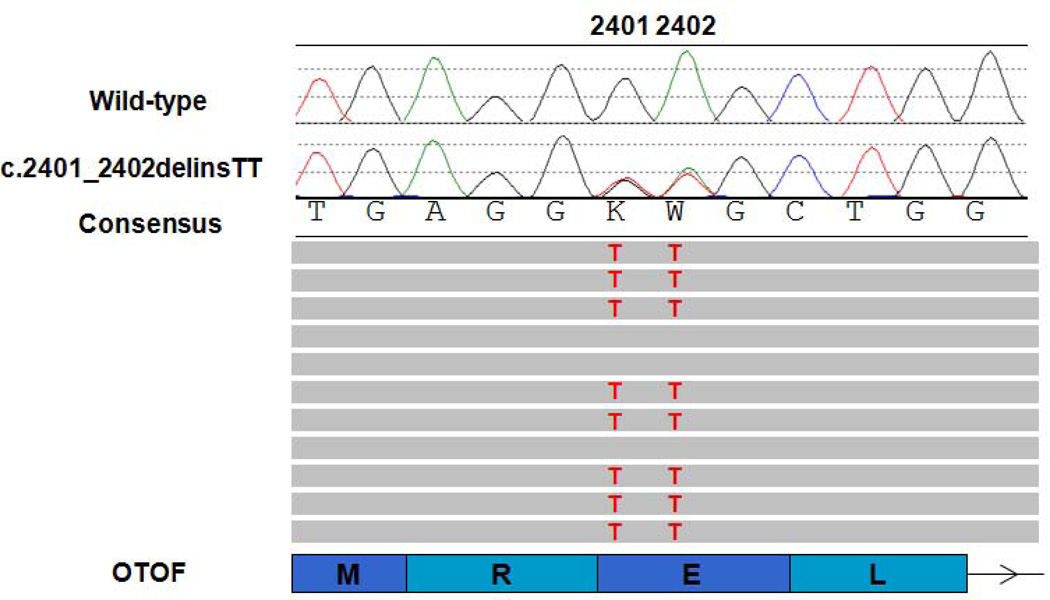
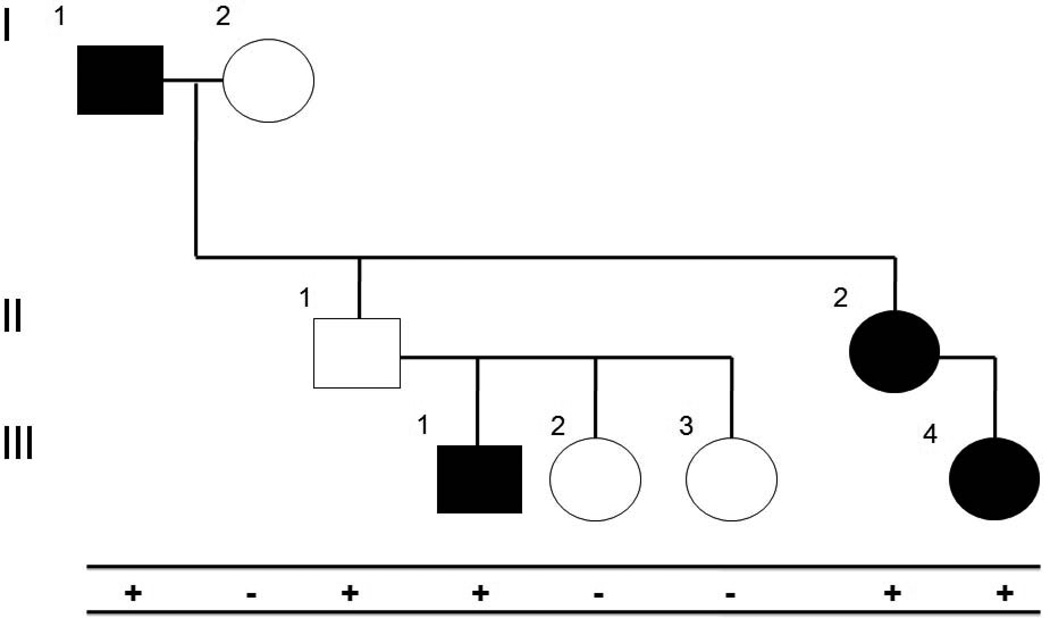
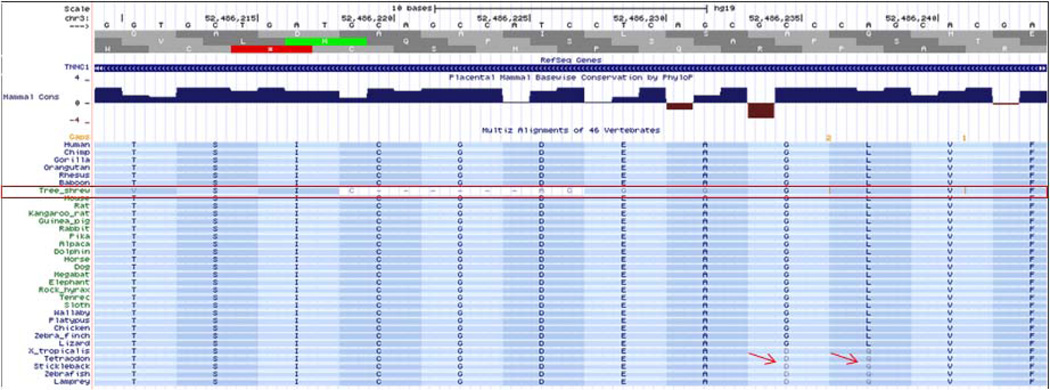
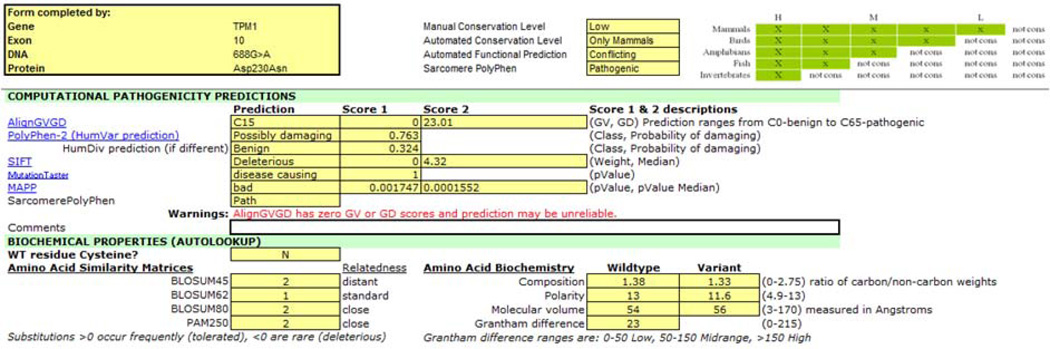
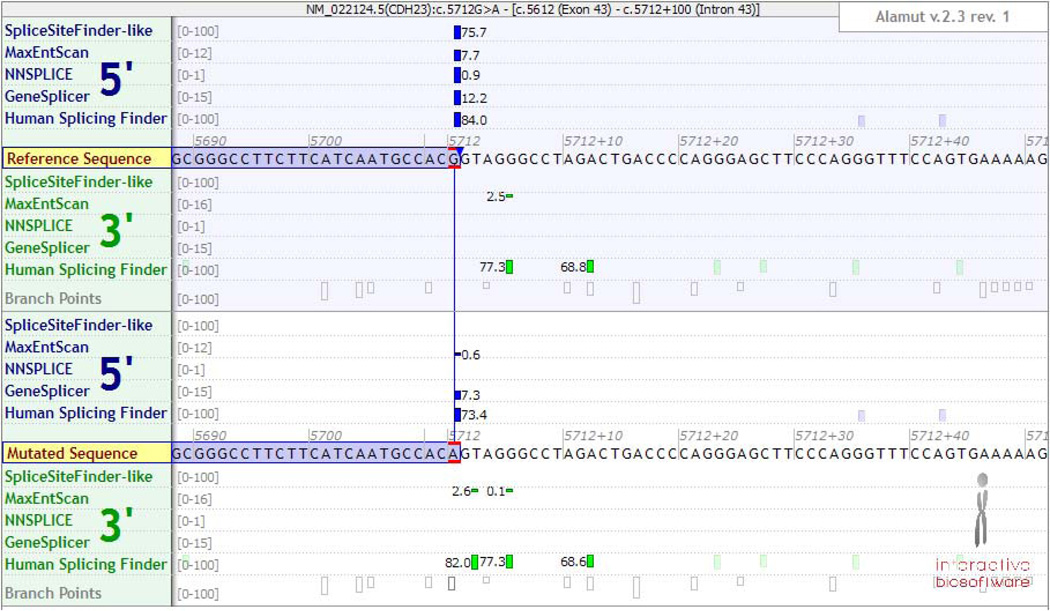
References
-
- Richards CS, Bradley LA, Amos J, Allitto B, Grody WW, Maddalena A, McGinnis MJ, Prior TW, Popovich BW, Watson MS, et al. Standards and guidelines for CFTR mutation testing. Genet Med. 2002;4:379–391. - PubMed
-
- Richards CS, Bale S, Bellissimo DB, Das S, Grody WW, Hegde MR, Lyon E, Ward BE. ACMG recommendations for standards for interpretation and reporting of sequence variations: Revisions 2007. Genet Med. 2008;10:294–300. - PubMed
-
- Kearney HM, Thorland EC, Brown KK, Quintero-Rivera F, South ST Working Group of the American College of Medical Genetics Laboratory Quality Assurance, C. American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet Med. 2011;13:680–685. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
