

Reducing assembly complexity of microbial genomes with single-molecule sequencing
- PMID: 24034426
- PMCID: PMC4053942
- DOI: 10.1186/gb-2013-14-9-r101
Reducing assembly complexity of microbial genomes with single-molecule sequencing
Abstract
Background: The short reads output by first- and second-generation DNA sequencing instruments cannot completely reconstruct microbial chromosomes. Therefore, most genomes have been left unfinished due to the significant resources required to manually close gaps in draft assemblies. Third-generation, single-molecule sequencing addresses this problem by greatly increasing sequencing read length, which simplifies the assembly problem.
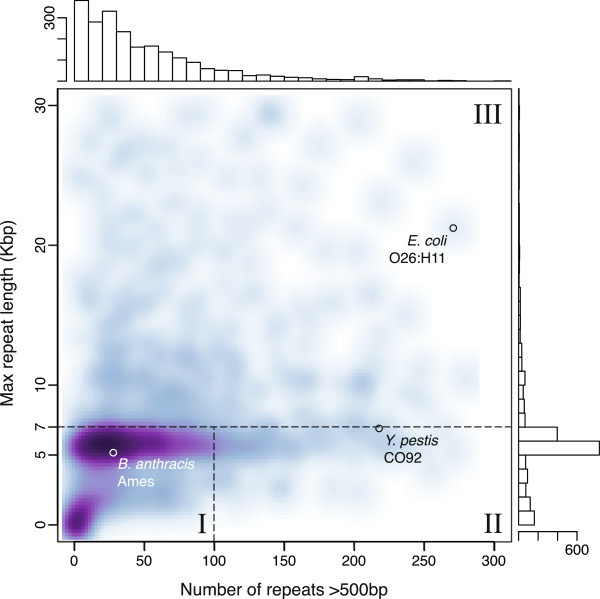
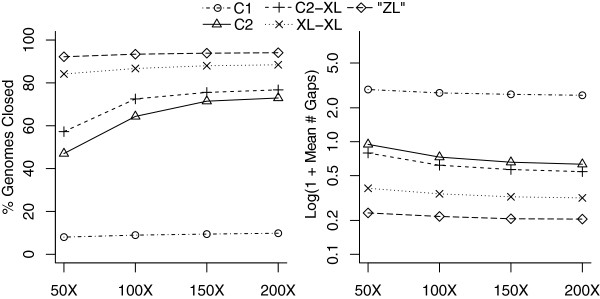
Results: To measure the benefit of single-molecule sequencing on microbial genome assembly, we sequenced and assembled the genomes of six bacteria and analyzed the repeat complexity of 2,267 complete bacteria and archaea. Our results indicate that the majority of known bacterial and archaeal genomes can be assembled without gaps, at finished-grade quality, using a single PacBio RS sequencing library. These single-library assemblies are also more accurate than typical short-read assemblies and hybrid assemblies of short and long reads.
Conclusions: Automated assembly of long, single-molecule sequencing data reduces the cost of microbial finishing to $1,000 for most genomes, and future advances in this technology are expected to drive the cost lower. This is expected to increase the number of completed genomes, improve the quality of microbial genome databases, and enable high-fidelity, population-scale studies of pan-genomes and chromosomal organization.
Figures
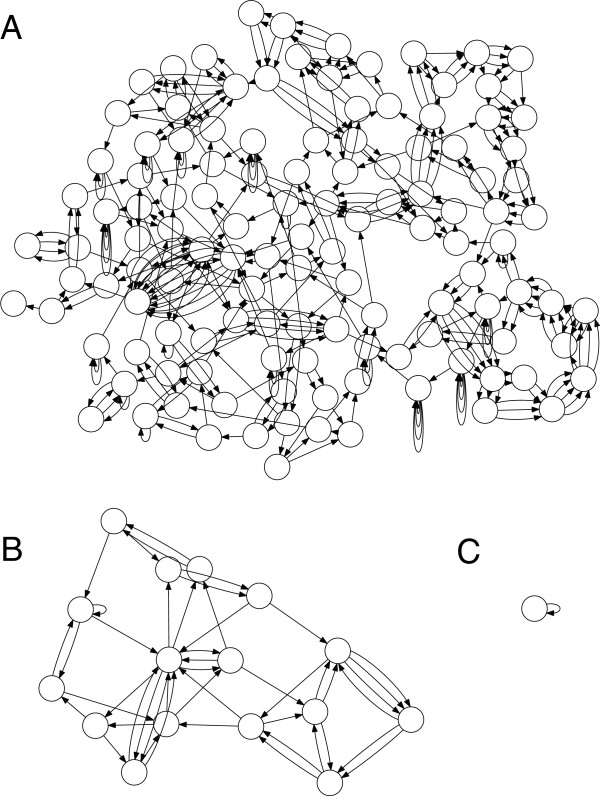
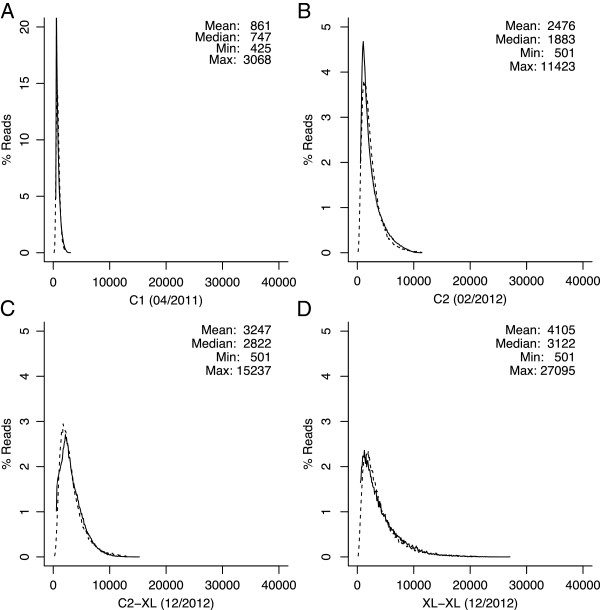
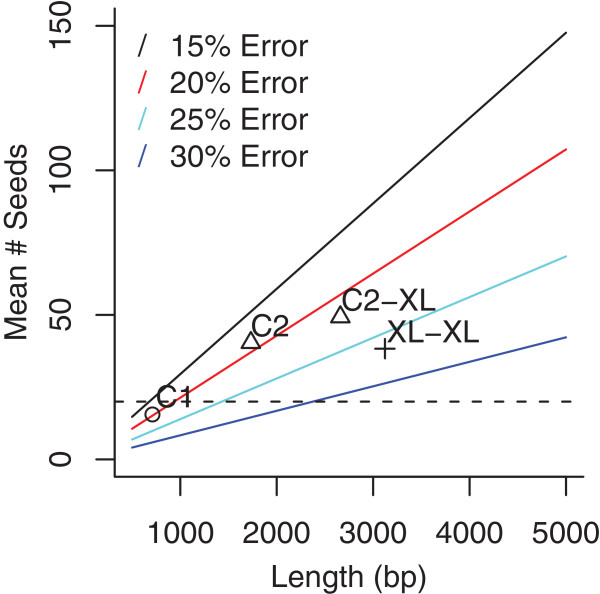
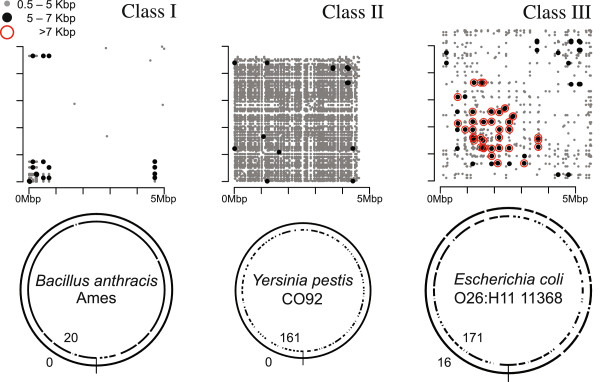
References
-
- Kyrpides NC. Genomes OnLine database (GOLD 1.0): A monitor of complete and ongoing genome projects world-wide. Bioinformatics. 1999;14:773–774. - PubMed
-
- Chain PS, Grafham DV, Fulton RS, Fitzgerald MG, Hostetler J, Muzny D, Ali J, Birren B, Bruce DC, Buhay C, Cole JR, Ding Y, Dugan S, Field D, Garrity GM, Gibbs R, Graves T, Han CS, Harrison SH, Highlander S, Hugenholtz P, Khouri HM, Kodira CD, Kolker E, Kyrpides NC, Lang D, Lapidus A, Malfatti SA, Markowitz V, Metha T. et al.Genomics. Genome project standards in a new era of sequencing. Science. 2009;14:236–237. - PMC - PubMed
-
- Salzberg SL, Yorke JA. Beware of mis-assembled genomes. Bioinformatics. 2005;14:4320–4321. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
