

Differential DNA methylation with age displays both common and dynamic features across human tissues that are influenced by CpG landscape
- PMID: 24034465
- PMCID: PMC4053985
- DOI: 10.1186/gb-2013-14-9-r102
Differential DNA methylation with age displays both common and dynamic features across human tissues that are influenced by CpG landscape
Abstract
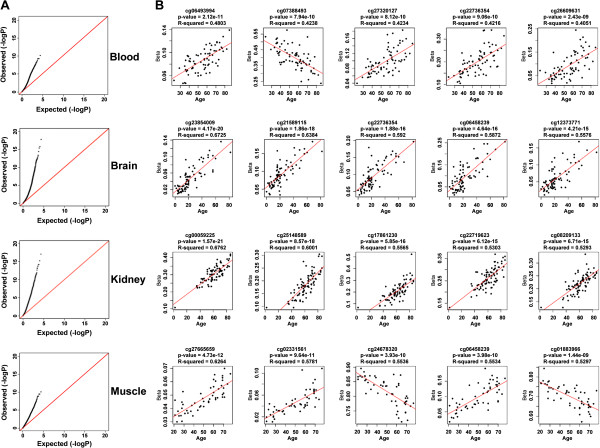
Background: DNA methylation is an epigenetic modification that changes with age in human tissues, although the mechanisms and specificity of this process are still poorly understood. We compared CpG methylation changes with age across 283 human blood, brain, kidney, and skeletal muscle samples using methylation arrays to identify tissue-specific age effects.
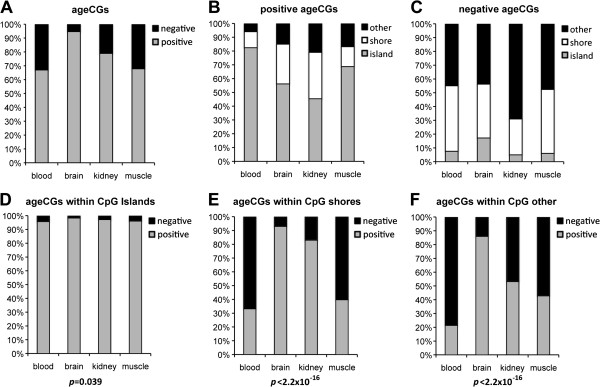
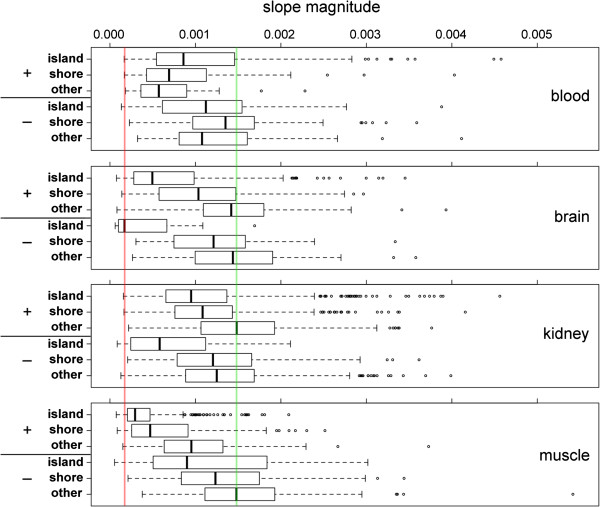
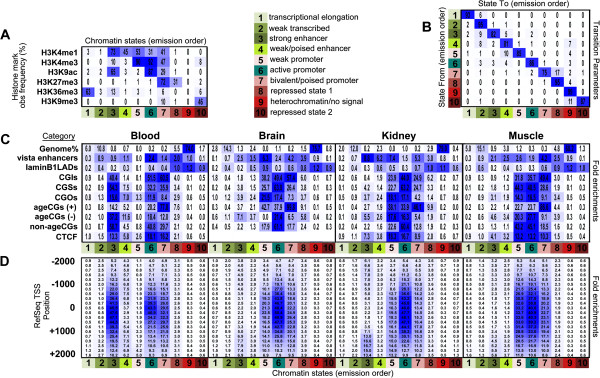
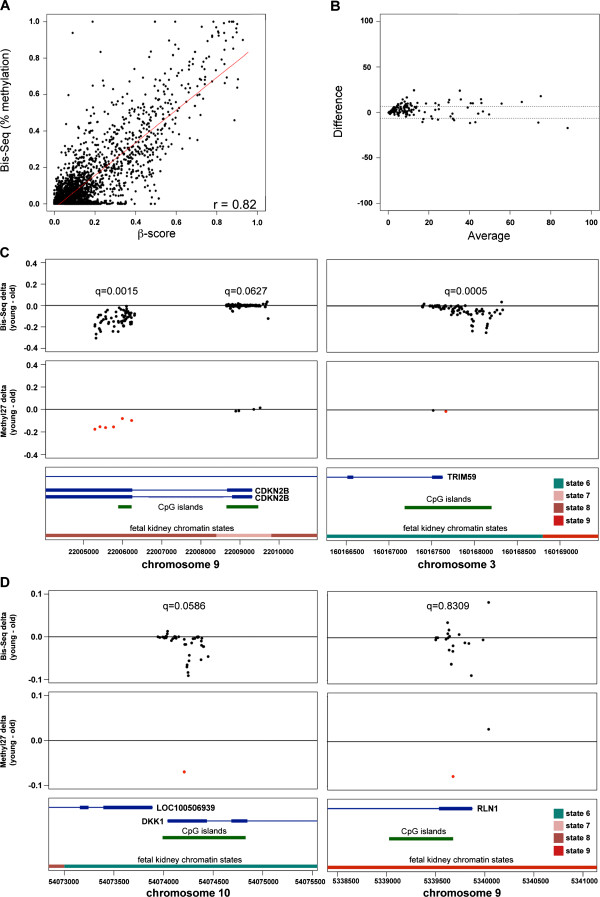
Results: We found age-associated CpGs (ageCGs) that are both tissue-specific and common across tissues. Tissue-specific age CGs are frequently located outside CpG islands with decreased methylation, and common ageCGs show the opposite trend. AgeCGs are significantly associated with poorly expressed genes, but those with decreasing methylation are linked with higher tissue-specific expression levels compared with increasing methylation. Therefore, tissue-specific gene expression may protect against common age-dependent methylation. Distinguished from other tissues, skeletal muscle age CGs are more associated with expression, enriched near genes related to myofiber contraction, and closer to muscle-specific CTCF binding sites. Kidney-specific ageCGs are more increasingly methylated compared to other tissues as measured by affiliation with kidney-specific expressed genes. Underlying chromatin features also mark common and tissue-specific age effects reflective of poised and active chromatin states, respectively. In contrast with decreasingly methylated ageCGs, increasingly methylated ageCGs are also generally further from CTCF binding sites and enriched within lamina associated domains.
Conclusions: Our data identified common and tissue-specific DNA methylation changes with age that are reflective of CpG landscape and suggests both common and unique alterations within human tissues. Our findings also indicate that a simple epigenetic drift model is insufficient to explain all age-related changes in DNA methylation.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
