

Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells
- PMID: 24036476
- PMCID: PMC3806088
- DOI: 10.1038/ncb2837
Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells
Abstract
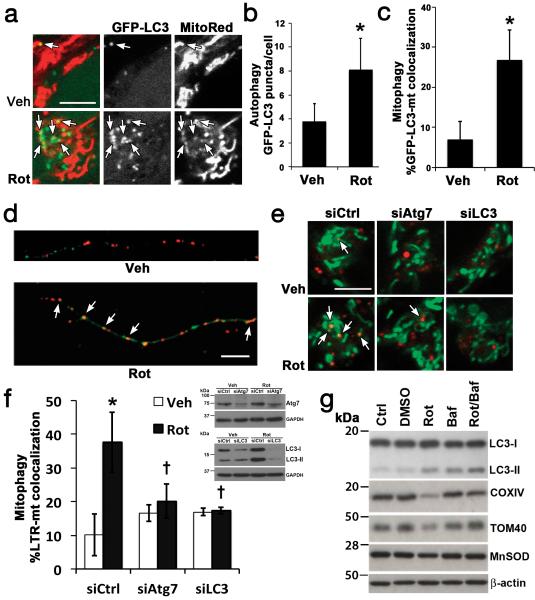
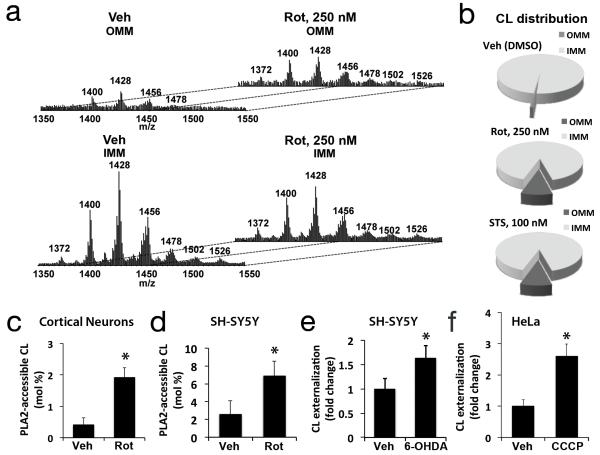
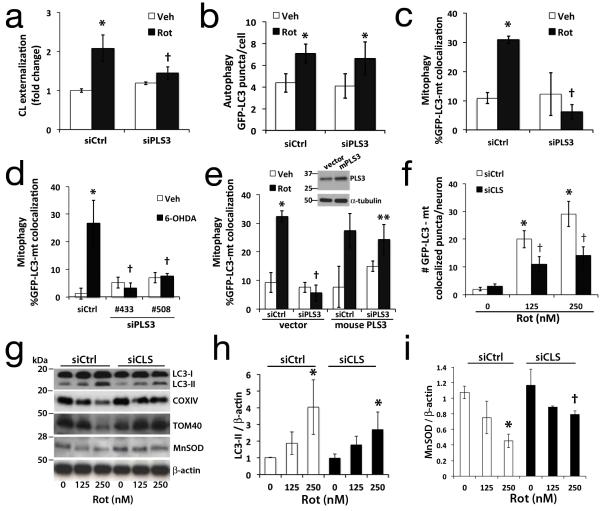
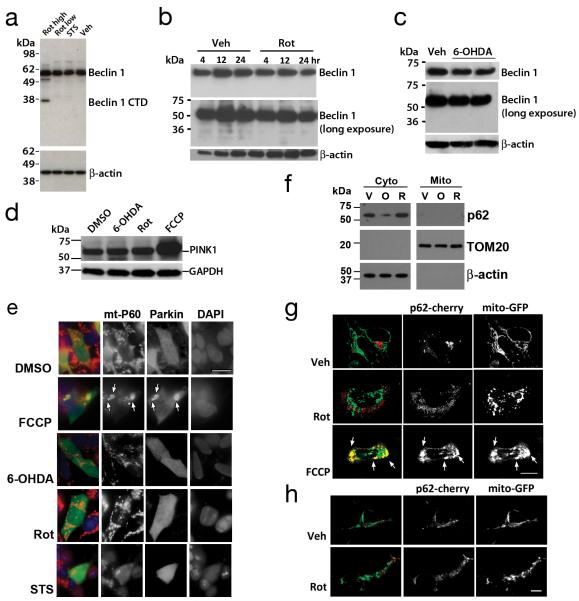
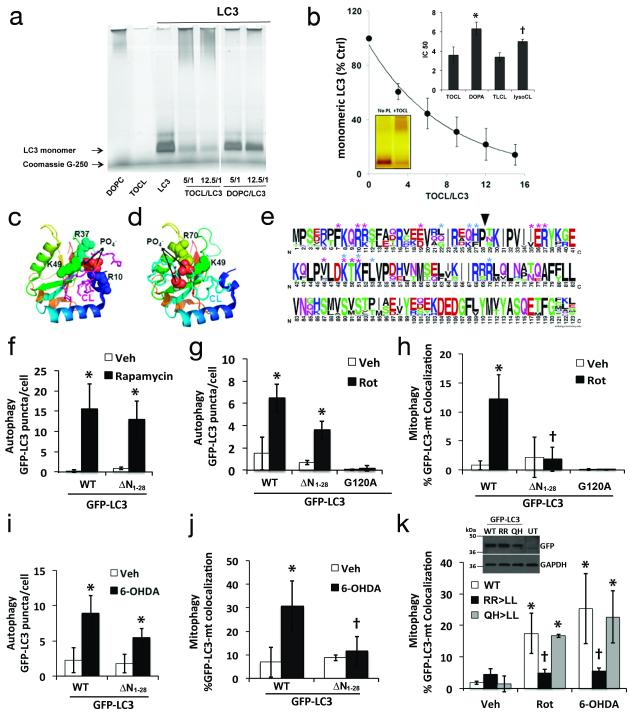
Recognition of injured mitochondria for degradation by macroautophagy is essential for cellular health, but the mechanisms remain poorly understood. Cardiolipin is an inner mitochondrial membrane phospholipid. We found that rotenone, staurosporine, 6-hydroxydopamine and other pro-mitophagy stimuli caused externalization of cardiolipin to the mitochondrial surface in primary cortical neurons and SH-SY5Y cells. RNAi knockdown of cardiolipin synthase or of phospholipid scramblase-3, which transports cardiolipin to the outer mitochondrial membrane, decreased the delivery of mitochondria to autophagosomes. Furthermore, we found that the autophagy protein microtubule-associated-protein-1 light chain 3 (LC3), which mediates both autophagosome formation and cargo recognition, contains cardiolipin-binding sites important for the engulfment of mitochondria by the autophagic system. Mutation of LC3 residues predicted as cardiolipin-interaction sites by computational modelling inhibited its participation in mitophagy. These data indicate that redistribution of cardiolipin serves as an 'eat-me' signal for the elimination of damaged mitochondria from neuronal cells.
Figures

References
-
- Liu L, et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxiainduced mitophagy in mammalian cells. Nature cell biology. 2012;14:177–185. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 HL070755/HL/NHLBI NIH HHS/United States
- R01 OH008282/OH/NIOSH CDC HHS/United States
- U19 AI068021/AI/NIAID NIH HHS/United States
- NS065789/NS/NINDS NIH HHS/United States
- AG026389/AG/NIA NIH HHS/United States
- R37 NS061817/NS/NINDS NIH HHS/United States
- R01 NS076511/NS/NINDS NIH HHS/United States
- ES020693/ES/NIEHS NIH HHS/United States
- R56 NS065789/NS/NINDS NIH HHS/United States
- F32 AG030821/AG/NIA NIH HHS/United States
- HL70755/HL/NHLBI NIH HHS/United States
- R01 AG026389/AG/NIA NIH HHS/United States
- F32AG030821/AG/NIA NIH HHS/United States
- R21 ES021068/ES/NIEHS NIH HHS/United States
- NS061817/NS/NINDS NIH HHS/United States
- R01 NS065789/NS/NINDS NIH HHS/United States
- R01 ES020693/ES/NIEHS NIH HHS/United States
- P41GM103712/GM/NIGMS NIH HHS/United States
- U19AIO68021/PHS HHS/United States
- R01 HL094488/HL/NHLBI NIH HHS/United States
- R21 HD057587/HD/NICHD NIH HHS/United States
- P20 GM103554/GM/NIGMS NIH HHS/United States
- NS076511/NS/NINDS NIH HHS/United States
- OH008282/OH/NIOSH CDC HHS/United States
- R01 NS061817/NS/NINDS NIH HHS/United States
- P41 GM103712/GM/NIGMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
