

Human gut microbiota community structures in urban and rural populations in Russia
- PMID: 24036685
- PMCID: PMC3778515
- DOI: 10.1038/ncomms3469
Human gut microbiota community structures in urban and rural populations in Russia
Abstract
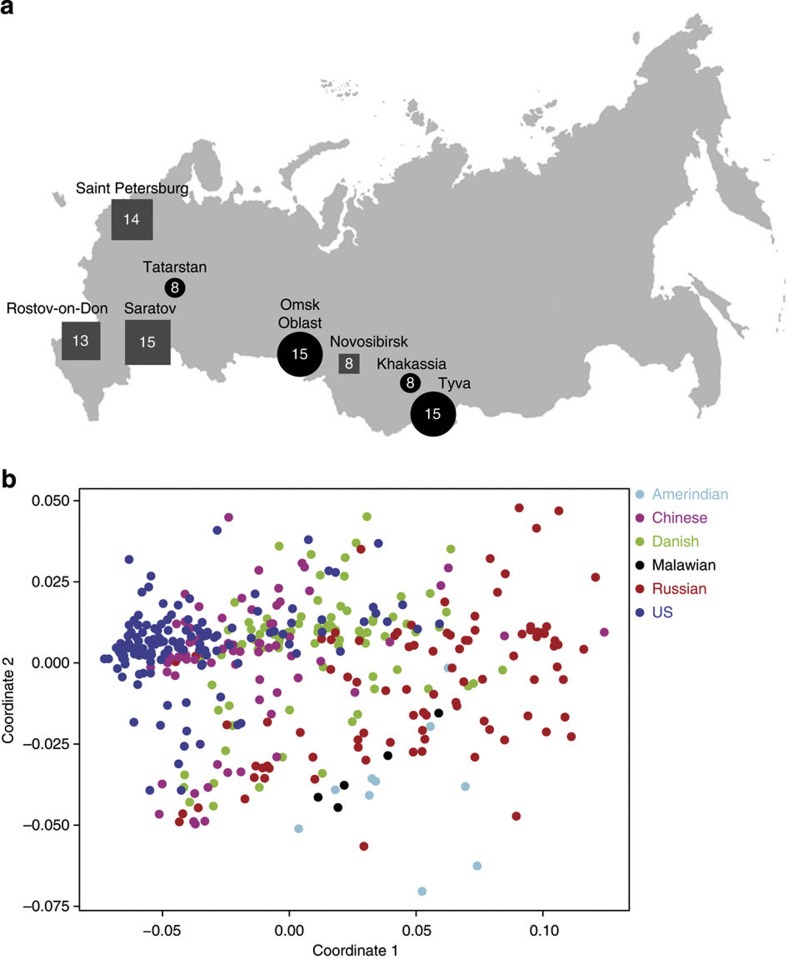
The microbial community of the human gut has a crucial role in sustaining host homeostasis. High-throughput DNA sequencing has delineated the structural and functional configurations of gut metagenomes in world populations. The microbiota of the Russian population is of particular interest to researchers, because Russia encompasses a uniquely wide range of environmental conditions and ethnogeographical cohorts. Here we conduct a shotgun metagenomic analysis of gut microbiota samples from 96 healthy Russian adult subjects, which reveals novel microbial community structures. The communities from several rural regions display similarities within each region and are dominated by the bacterial taxa associated with the healthy gut. Functional analysis shows that the metabolic pathways exhibiting differential abundance in the novel types are primarily associated with the trade-off between the Bacteroidetes and Firmicutes phyla. The specific signatures of the Russian gut microbiota are likely linked to the host diet, cultural habits and socioeconomic status.
Figures




References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
