

Network-based stratification of tumor mutations
- PMID: 24037242
- PMCID: PMC3866081
- DOI: 10.1038/nmeth.2651
Network-based stratification of tumor mutations
Abstract
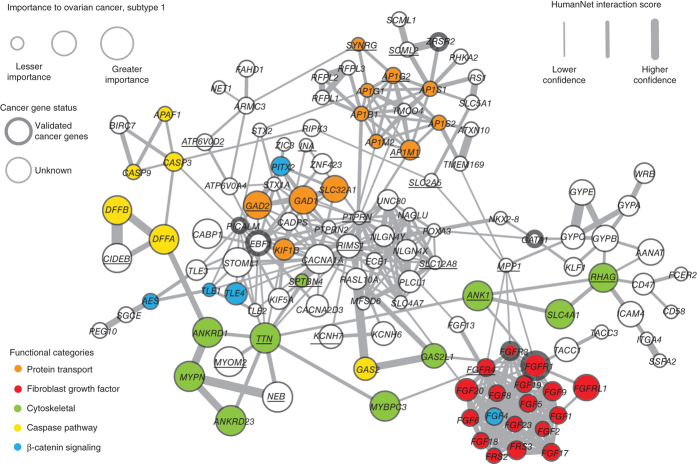
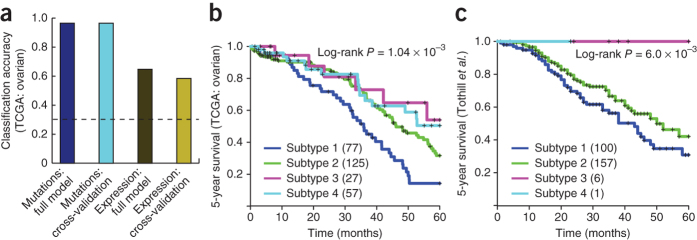
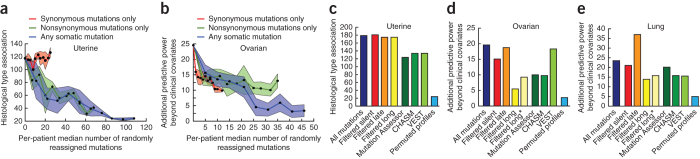
Many forms of cancer have multiple subtypes with different causes and clinical outcomes. Somatic tumor genome sequences provide a rich new source of data for uncovering these subtypes but have proven difficult to compare, as two tumors rarely share the same mutations. Here we introduce network-based stratification (NBS), a method to integrate somatic tumor genomes with gene networks. This approach allows for stratification of cancer into informative subtypes by clustering together patients with mutations in similar network regions. We demonstrate NBS in ovarian, uterine and lung cancer cohorts from The Cancer Genome Atlas. For each tissue, NBS identifies subtypes that are predictive of clinical outcomes such as patient survival, response to therapy or tumor histology. We identify network regions characteristic of each subtype and show how mutation-derived subtypes can be used to train an mRNA expression signature, which provides similar information in the absence of DNA sequence.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
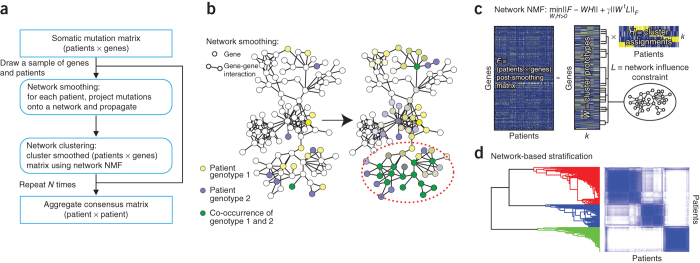
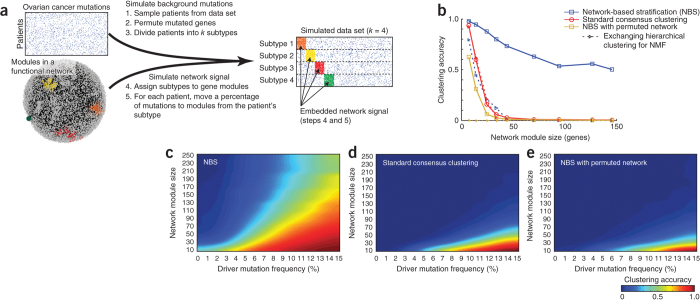
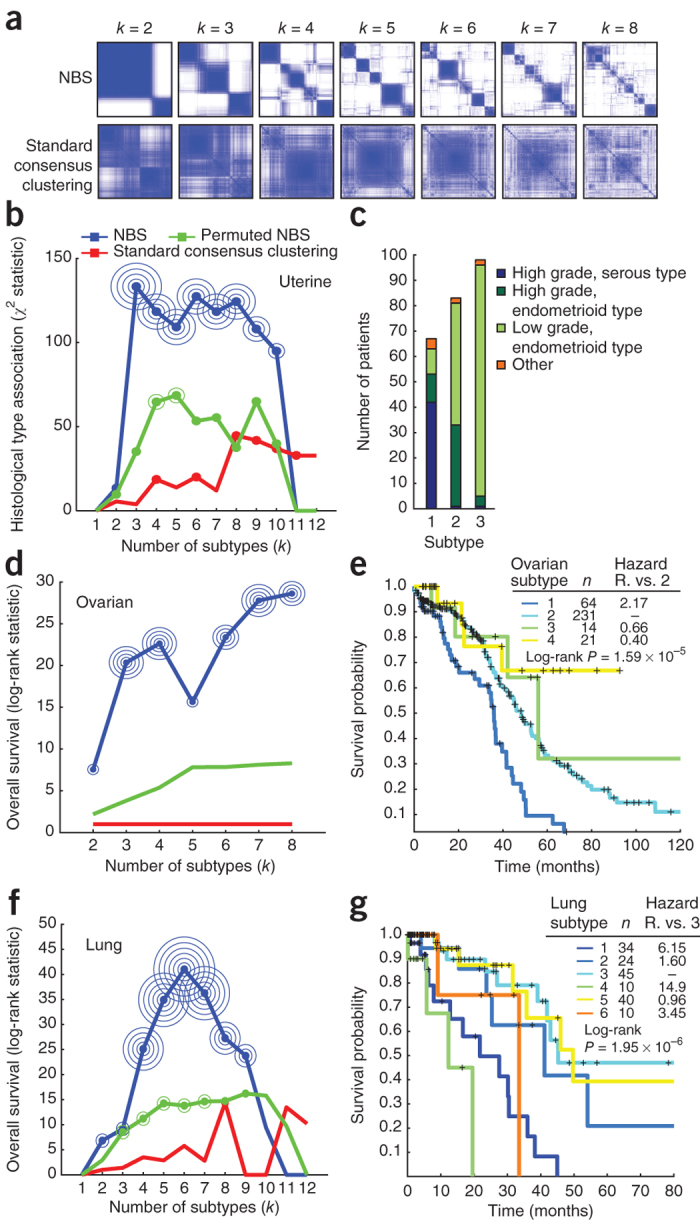
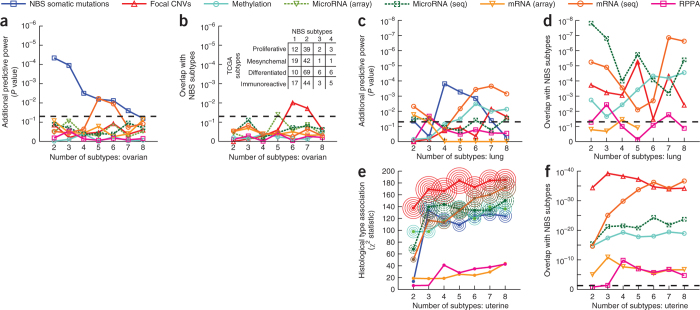
Comment in
-
Making connections: using networks to stratify human tumors.Nat Methods. 2013 Nov;10(11):1077-8. doi: 10.1038/nmeth.2704. Nat Methods. 2013. PMID: 24173383 No abstract available.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
