

Relating phylogenetic trees to transmission trees of infectious disease outbreaks
- PMID: 24037268
- PMCID: PMC3813836
- DOI: 10.1534/genetics.113.154856
Relating phylogenetic trees to transmission trees of infectious disease outbreaks
Abstract
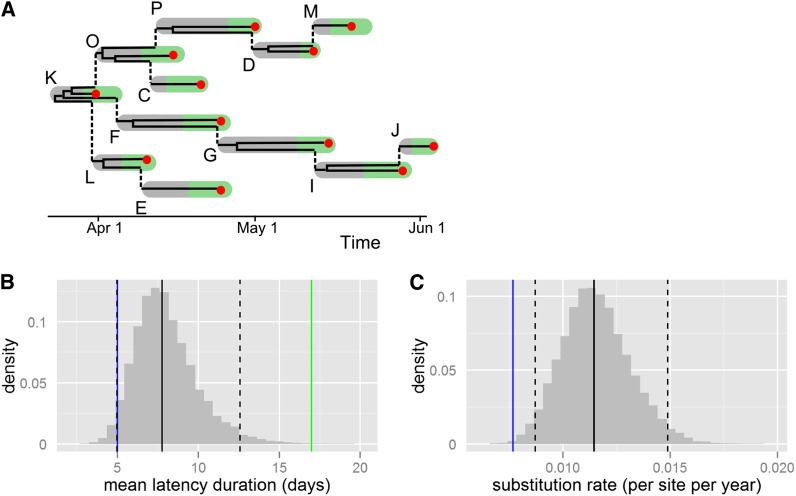
Transmission events are the fundamental building blocks of the dynamics of any infectious disease. Much about the epidemiology of a disease can be learned when these individual transmission events are known or can be estimated. Such estimations are difficult and generally feasible only when detailed epidemiological data are available. The genealogy estimated from genetic sequences of sampled pathogens is another rich source of information on transmission history. Optimal inference of transmission events calls for the combination of genetic data and epidemiological data into one joint analysis. A key difficulty is that the transmission tree, which describes the transmission events between infected hosts, differs from the phylogenetic tree, which describes the ancestral relationships between pathogens sampled from these hosts. The trees differ both in timing of the internal nodes and in topology. These differences become more pronounced when a higher fraction of infected hosts is sampled. We show how the phylogenetic tree of sampled pathogens is related to the transmission tree of an outbreak of an infectious disease, by the within-host dynamics of pathogens. We provide a statistical framework to infer key epidemiological and mutational parameters by simultaneously estimating the phylogenetic tree and the transmission tree. We test the approach using simulations and illustrate its use on an outbreak of foot-and-mouth disease. The approach unifies existing methods in the emerging field of phylodynamics with transmission tree reconstruction methods that are used in infectious disease epidemiology.
Keywords: Markov chain Monte Carlo (MCMC); foot-and-mouth disease; molecular epidemiology; transmission tree.
Figures



References
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
