

Position of glycine substitutions in the triple helix of COL6A1, COL6A2, and COL6A3 is correlated with severity and mode of inheritance in collagen VI myopathies
- PMID: 24038877
- PMCID: PMC4520221
- DOI: 10.1002/humu.22429
Position of glycine substitutions in the triple helix of COL6A1, COL6A2, and COL6A3 is correlated with severity and mode of inheritance in collagen VI myopathies
Abstract
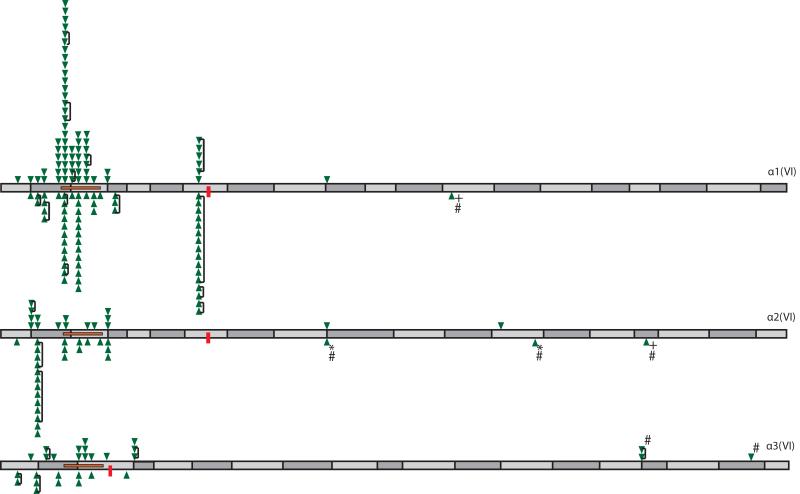
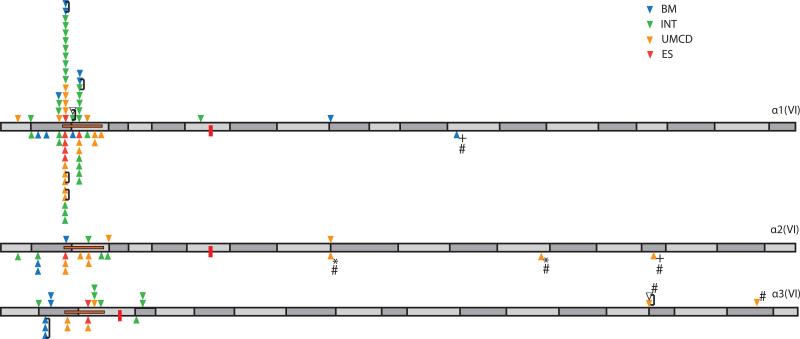
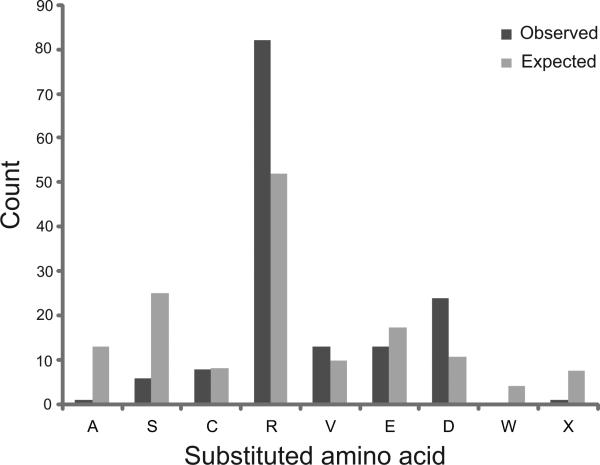
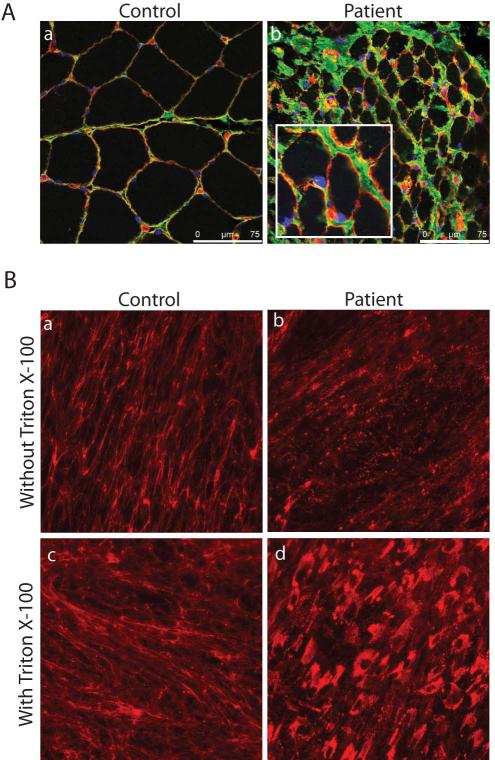
Glycine substitutions in the conserved Gly-X-Y motif in the triple helical (TH) domain of collagen VI are the most commonly identified mutations in the collagen VI myopathies including Ullrich congenital muscular dystrophy, Bethlem myopathy, and intermediate (INT) phenotypes. We describe clinical and genetic characteristics of 97 individuals with glycine substitutions in the TH domain of COL6A1, COL6A2, or COL6A3 and add a review of 97 published cases, for a total of 194 cases. Clinical findings include severe, INT, and mild phenotypes even from patients with identical mutations. INT phenotypes were most common, accounting for almost half of patients, emphasizing the importance of INT phenotypes to the overall phenotypic spectrum. Glycine substitutions in the TH domain are heavily clustered in a short segment N-terminal to the 17th Gly-X-Y triplet, where they are acting as dominants. The most severe cases are clustered in an even smaller region including Gly-X-Y triplets 10-15, accounting for only 5% of the TH domain. Our findings suggest that clustering of glycine substitutions in the N-terminal region of collagen VI is not based on features of the primary sequence. We hypothesize that this region may represent a functional domain within the triple helix.
Keywords: Bethlem myopathy; Ullrich congenital muscular dystrophy; collagen VI; genotype-phenotype correlation.
Published 2013. Wiley Periodicals, Inc. *This article is a U.S. Government work and is in the public domain in the USA.
Figures


References
-
- Baker NL, Morgelin M, Pace RA, Peat RA, Adams NE, Gardner RJ, Rowland LP, Miller G, De Jonghe P, Ceulemans B. Molecular consequences of dominant Bethlem myopathy collagen VI mutations. Ann Neurol. 2007;62(4):390–405. others. - PubMed
-
- Baker NL, Morgelin M, Peat R, Goemans N, North KN, Bateman JF, Lamande SR. Dominant collagen VI mutations are a common cause of Ullrich congenital muscular dystrophy. Hum Mol Genet. 2005;14(2):279–93. - PubMed
-
- Baldock C, Sherratt MJ, Shuttleworth CA, Kielty CM. The supramolecular organization of collagen VI microfibrils. J Mol Biol. 2003;330(2):297–307. - PubMed
-
- Brinas L, Richard P, Quijano-Roy S, Gartioux C, Ledeuil C, Lacene E, Makri S, Ferreiro A, Maugenre S, Topaloglu H. Early onset collagen VI myopathies: Genetic and clinical correlations. Ann Neurol. 2010;68(4):511–20. others. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
