

Mutation spectrum and genotype-phenotype correlation in Cornelia de Lange syndrome
- PMID: 24038889
- PMCID: PMC3880228
- DOI: 10.1002/humu.22430
Mutation spectrum and genotype-phenotype correlation in Cornelia de Lange syndrome
Abstract
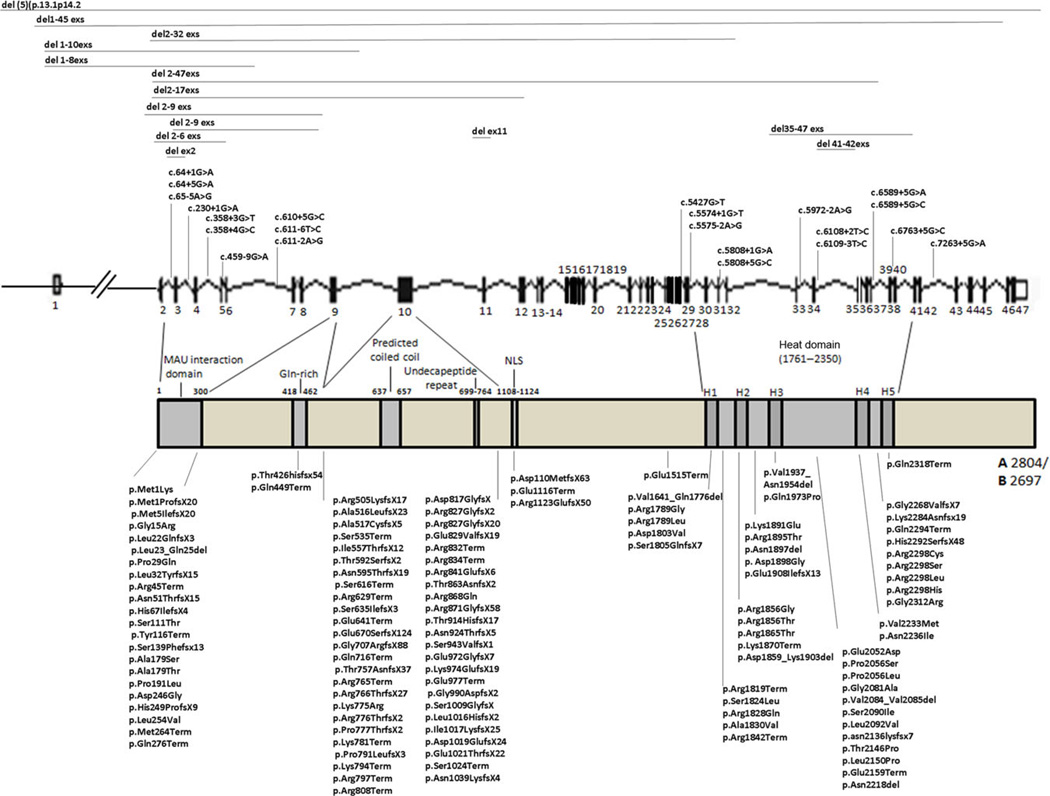
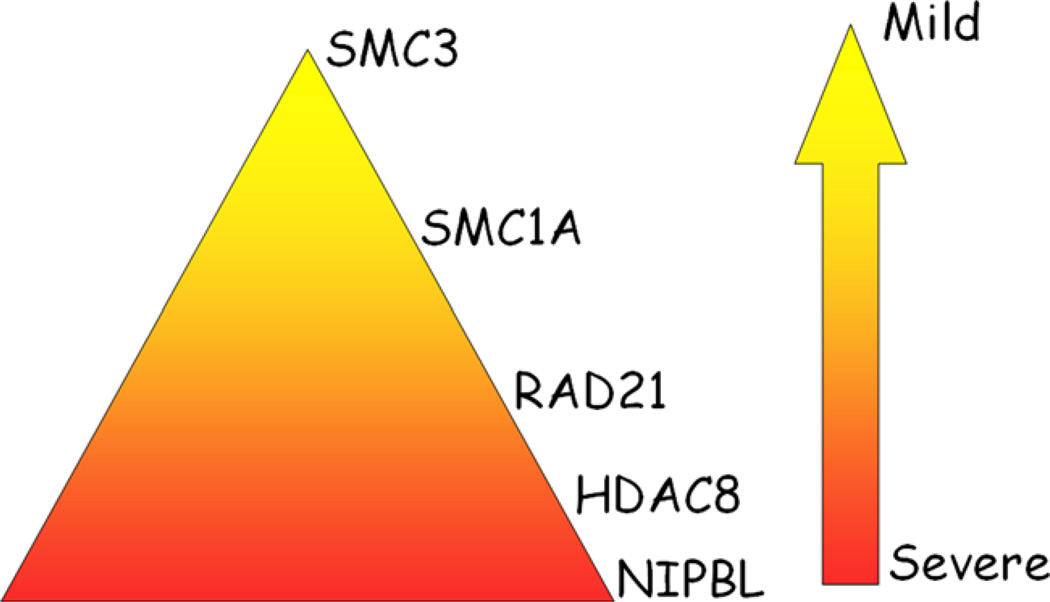
Cornelia de Lange syndrome (CdLS) is a clinically and genetically heterogeneous developmental disorder. Clinical features include growth retardation, intellectual disability, limb defects, typical facial dysmorphism, and other systemic involvement. The increased understanding of the genetic basis of CdLS has led to diagnostic improvement and expansion of the phenotype. Mutations in five genes (NIPBL, SMC1A, SMC3, RAD21, and HDAC8), all regulators or structural components of cohesin, have been identified. Approximately 60% of CdLS cases are due to NIPBL mutations, 5% caused by mutations in SMC1A, RAD21, and HDAC8 and one proband was found to carry a mutation in SMC3. To date, 311 CdLS-causing mutations are known including missense, nonsense, small deletions and insertions, splice site mutations, and genomic rearrangements. Phenotypic variability is seen both intra- and intergenically. This article reviews the spectrum of CdLS mutations with a particular emphasis on their correlation to the clinical phenotype.
Keywords: Cornelia de Lange syndrome; HDAC8; NIPBL; RAD21; SMC1A; SMC3.
© 2013 WILEY PERIODICALS, INC.
Conflict of interest statement
Figures


References
-
- Borck G, Zarhrate M, Cluzeau C, Bal E, Bonnefont JP, Munnich A, Cormier-Daire V, Colleaux L. Father-to-daughter transmission of Cornelia de Lange syndrome caused by a mutation in the 5 ’ untranslated region of the NIPBL gene. Hum Mutat. 2006;27:731–735. - PubMed
-
- Brachmann W. Ein fall von symmetrischer monodaktylie durch Ulnadefekt, mit symmetrischer flughautbildung in den ellenbeugen, sowie anderen abnoanderen (zwerghaftogkeit, halsrippen, behaarung) Jarb Kinder Phys Erzie. 1916;84:225–235.
-
- Brown CJ, Miller AP, Carrel L, Rupert JL, Davies KE, Willard HF. The DXS423E gene inXp11.21 escapes Xchromosome inactivation. Hum Mol Genet. 1995;4:251–255. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
