

Genome analysis of a transmissible lineage of pseudomonas aeruginosa reveals pathoadaptive mutations and distinct evolutionary paths of hypermutators
- PMID: 24039595
- PMCID: PMC3764201
- DOI: 10.1371/journal.pgen.1003741
Genome analysis of a transmissible lineage of pseudomonas aeruginosa reveals pathoadaptive mutations and distinct evolutionary paths of hypermutators
Abstract
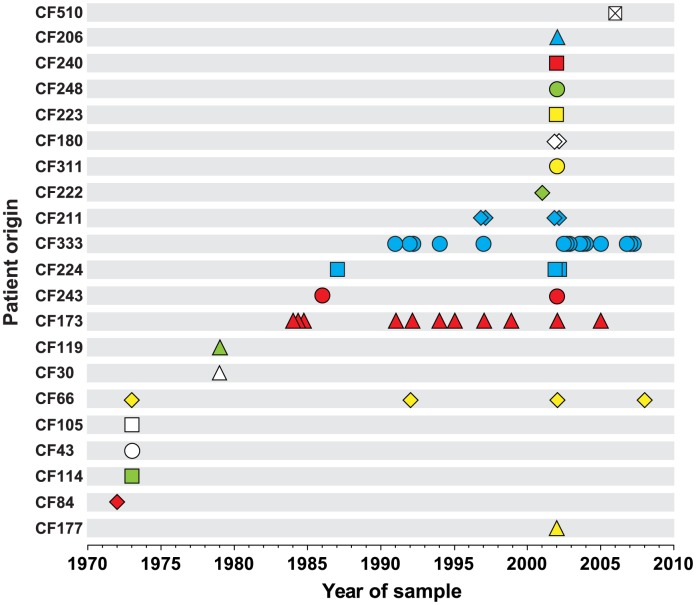
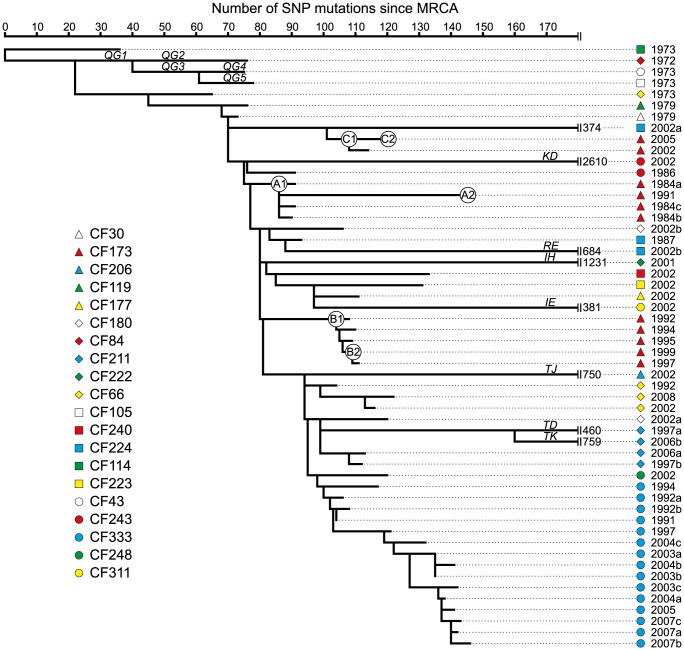
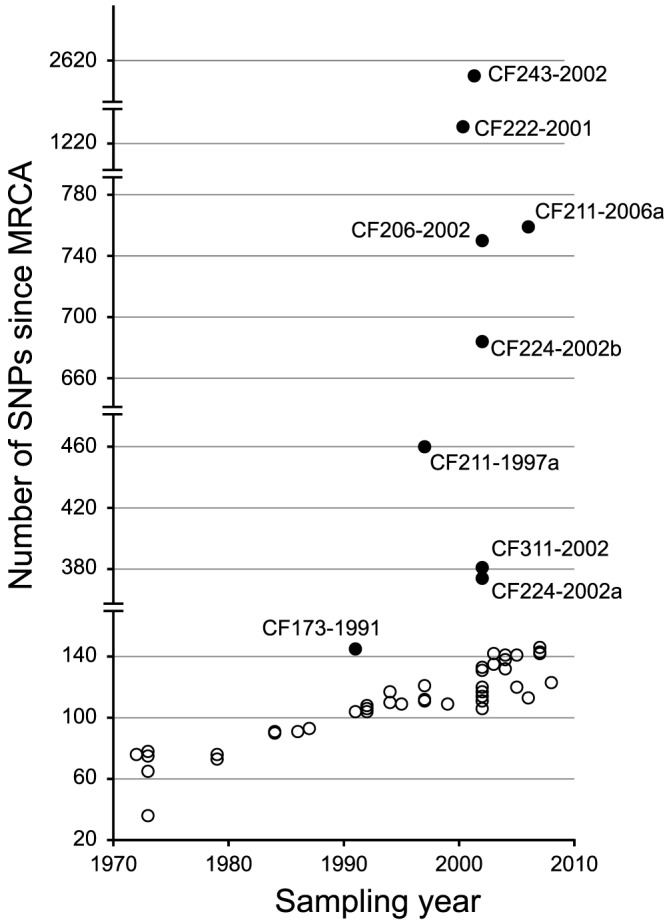
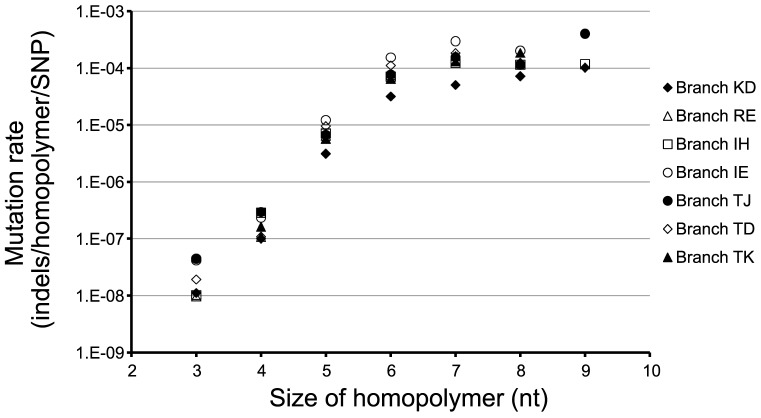
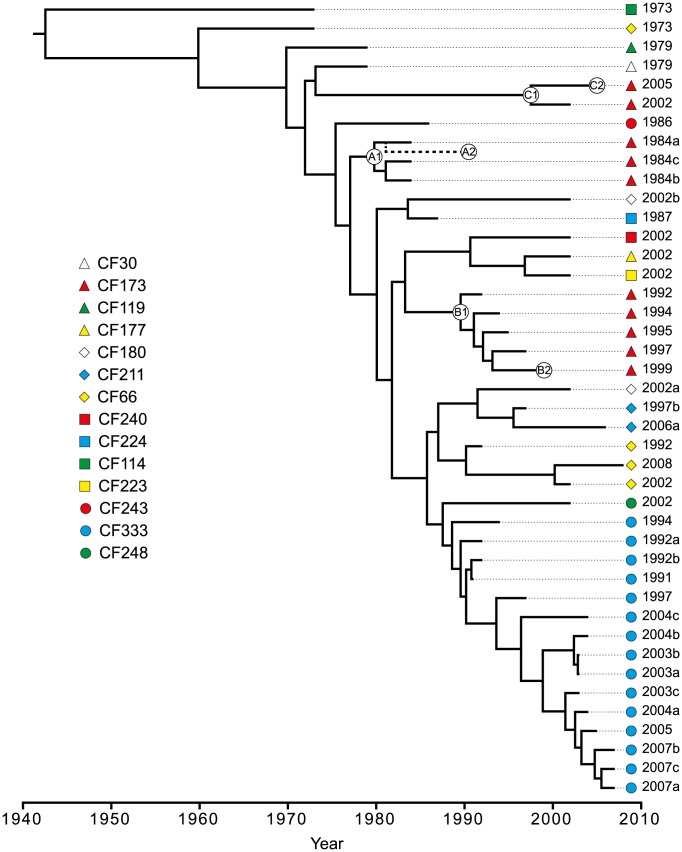
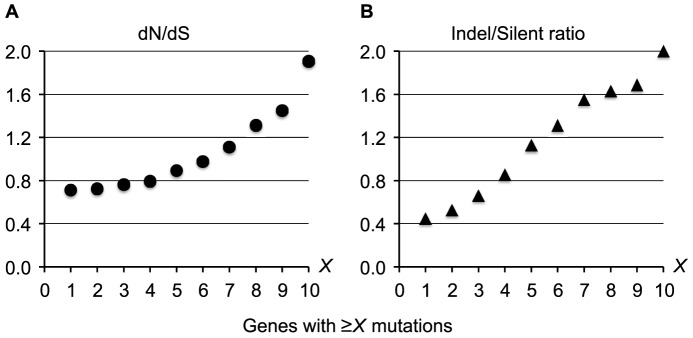
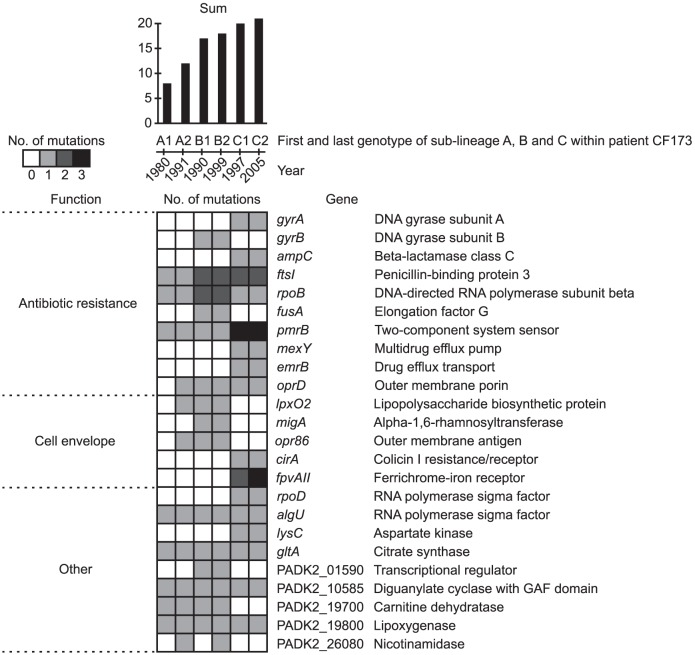
Genome sequencing of bacterial pathogens has advanced our understanding of their evolution, epidemiology, and response to antibiotic therapy. However, we still have only a limited knowledge of the molecular changes in in vivo evolving bacterial populations in relation to long-term, chronic infections. For example, it remains unclear what genes are mutated to facilitate the establishment of long-term existence in the human host environment, and in which way acquisition of a hypermutator phenotype with enhanced rates of spontaneous mutations influences the evolutionary trajectory of the pathogen. Here we perform a retrospective study of the DK2 clone type of P. aeruginosa isolated from Danish patients suffering from cystic fibrosis (CF), and analyze the genomes of 55 bacterial isolates collected from 21 infected individuals over 38 years. Our phylogenetic analysis of 8,530 mutations in the DK2 genomes shows that the ancestral DK2 clone type spread among CF patients through several independent transmission events. Subsequent to transmission, sub-lineages evolved independently for years in separate hosts, creating a unique possibility to study parallel evolution and identification of genes targeted by mutations to optimize pathogen fitness (pathoadaptive mutations). These genes were related to antibiotic resistance, the cell envelope, or regulatory functions, and we find that the prevalence of pathoadaptive mutations correlates with evolutionary success of co-evolving sub-lineages. The long-term co-existence of both normal and hypermutator populations enabled comparative investigations of the mutation dynamics in homopolymeric sequences in which hypermutators are particularly prone to mutations. We find a positive exponential correlation between the length of the homopolymer and its likelihood to acquire mutations and identify two homopolymer-containing genes preferentially mutated in hypermutators. This homopolymer facilitated differential mutagenesis provides a novel genome-wide perspective on the different evolutionary trajectories of hypermutators, which may help explain their emergence in CF infections.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
