

RBFOX1 and RBFOX3 mutations in rolandic epilepsy
- PMID: 24039908
- PMCID: PMC3765197
- DOI: 10.1371/journal.pone.0073323
RBFOX1 and RBFOX3 mutations in rolandic epilepsy
Abstract
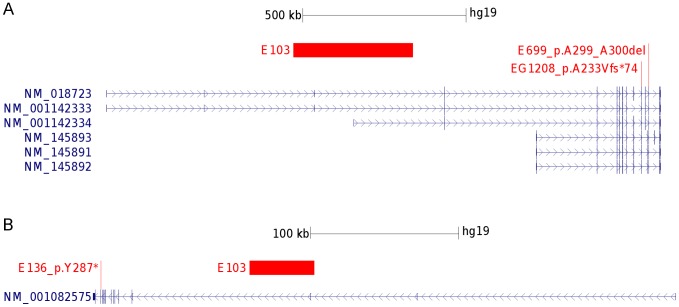
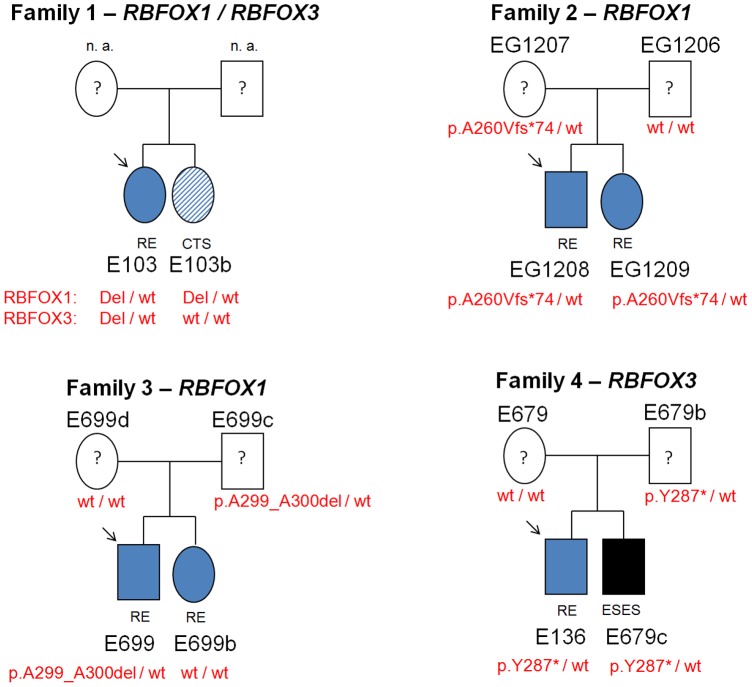
Partial deletions of the gene encoding the neuronal splicing regulator RBFOX1 have been reported in a range of neurodevelopmental diseases, including idiopathic generalized epilepsy. The RBFOX1 protein and its homologues (RBFOX2 and RBFOX3) regulate alternative splicing of many neuronal transcripts involved in the homeostatic control of neuronal excitability. In this study, we explored if structural microdeletions and exonic sequence variations in RBFOX1, RBFOX2, RBFOX3 confer susceptibility to rolandic epilepsy (RE), a common idiopathic focal childhood epilepsy. By high-density SNP array screening of 289 unrelated RE patients, we identified two hemizygous deletions, a 365 kb deletion affecting two untranslated 5'-terminal exons of RBFOX1 and a 43 kb deletion spanning exon 3 of RBFOX3. Exome sequencing of 242 RE patients revealed two novel probably deleterious variants in RBFOX1, a frameshift mutation (p.A233Vfs*74) and a hexanucleotide deletion (p.A299_A300del), and a novel nonsense mutation in RBFOX3 (p.Y287*). Although the three variants were inherited from unaffected parents, they were present in all family members exhibiting the RE trait clinically or electroencephalographically with only one exception. In contrast, no deleterious mutations of RBFOX1 and RBFOX3 were found in the exomes of 6503 non-RE subjects deposited in the Exome Variant Server database. The observed RBFOX3 exon 3 deletion and nonsense mutation suggest that RBFOX3 represents a novel risk factor for RE, indicating that exon deletions and truncating mutations of RBFOX1 and RBFOX3 contribute to the genetic variance of partial and generalized idiopathic epilepsy syndromes.
Conflict of interest statement
Figures
References
-
- Sidenvall R, Forsgren L, Blomquist HK, Heijbel J (1993) A community-based prospective incidence study of epileptic seizures in children. Acta Paediatr 82: 60–65. - PubMed
-
- Doose H, Brigger-Heuer B, Neubauer B (1997) Children with focal sharp waves: clinical and genetic aspects. Epilepsia 38: 788–796. - PubMed
-
- Berg AT, Berkovic SF, Brodie MJ, Buchhalter J, Cross JH, et al. (2010) Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia 51: 676–685 doi:10.1111/j.1528-1167.2010.02522.x - DOI - PubMed
-
- Ottman R (1989) Genetics of the partial epilepsies: a review. Epilepsia 30: 107–111. - PubMed
-
- Bali B, Kull LL, Strug LJ, Clarke T, Murphy PL, et al. (2007) Autosomal dominant inheritance of centrotemporal sharp waves in rolandic epilepsy families. Epilepsia 48: 2266–2272 doi:10.1111/j.1528-1167.2007.01221.x - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
