

Phosphofructokinase deficiency impairs ATP generation, autophagy, and redox balance in rheumatoid arthritis T cells
- PMID: 24043759
- PMCID: PMC3782046
- DOI: 10.1084/jem.20130252
Phosphofructokinase deficiency impairs ATP generation, autophagy, and redox balance in rheumatoid arthritis T cells
Abstract
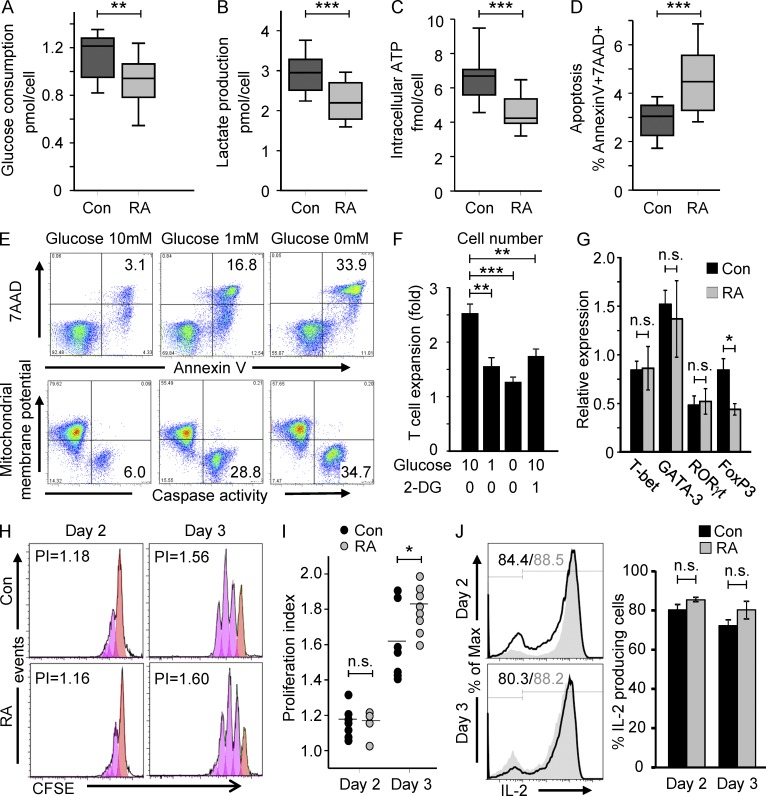
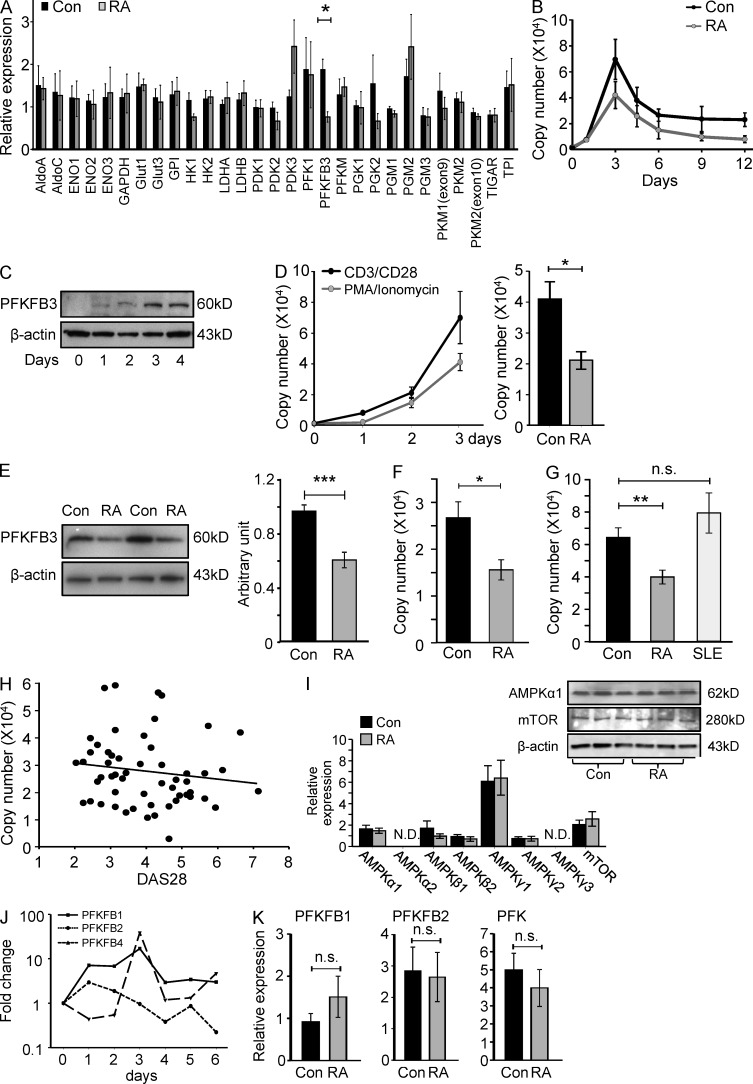
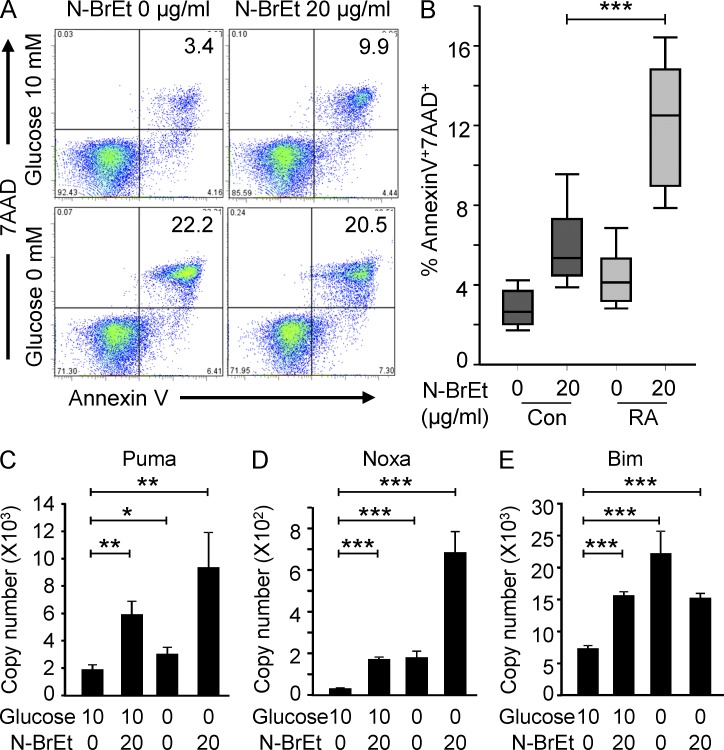
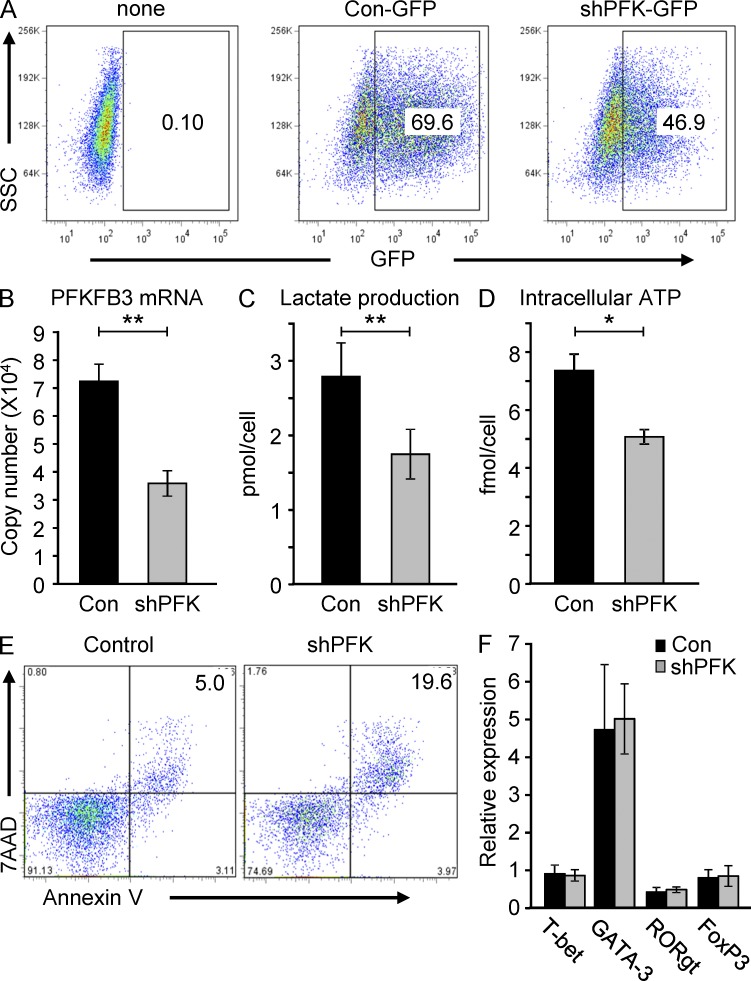
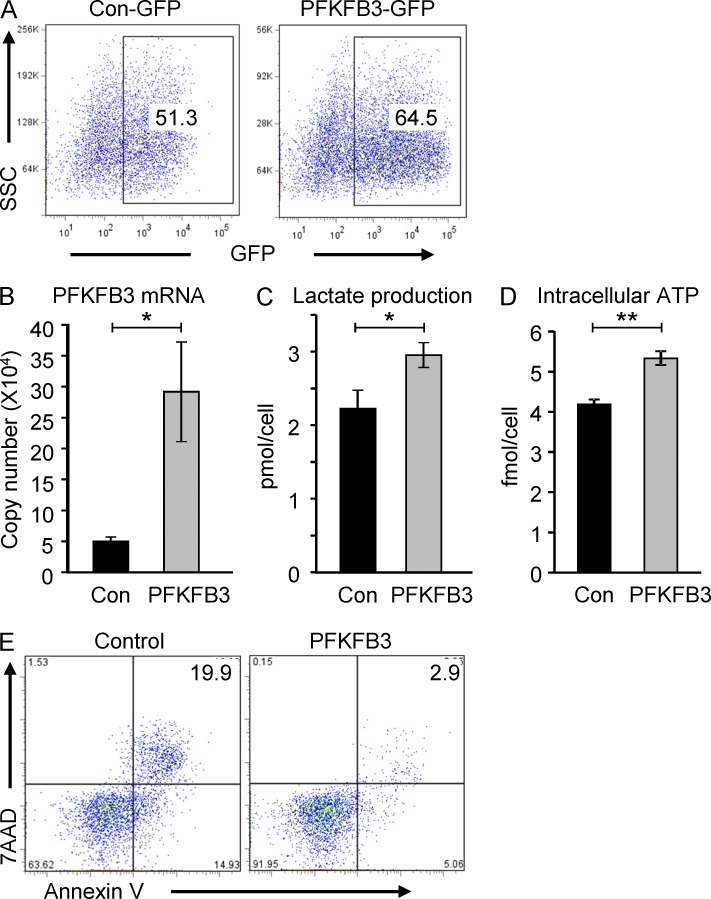
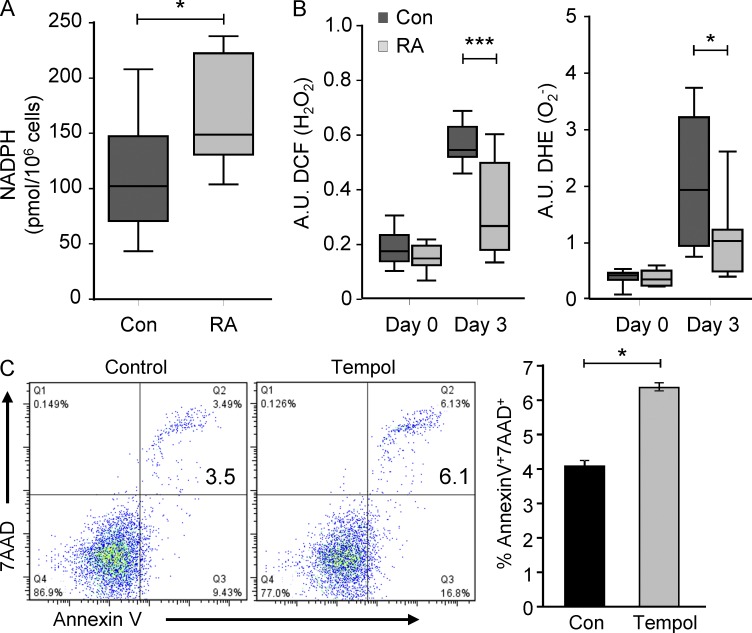
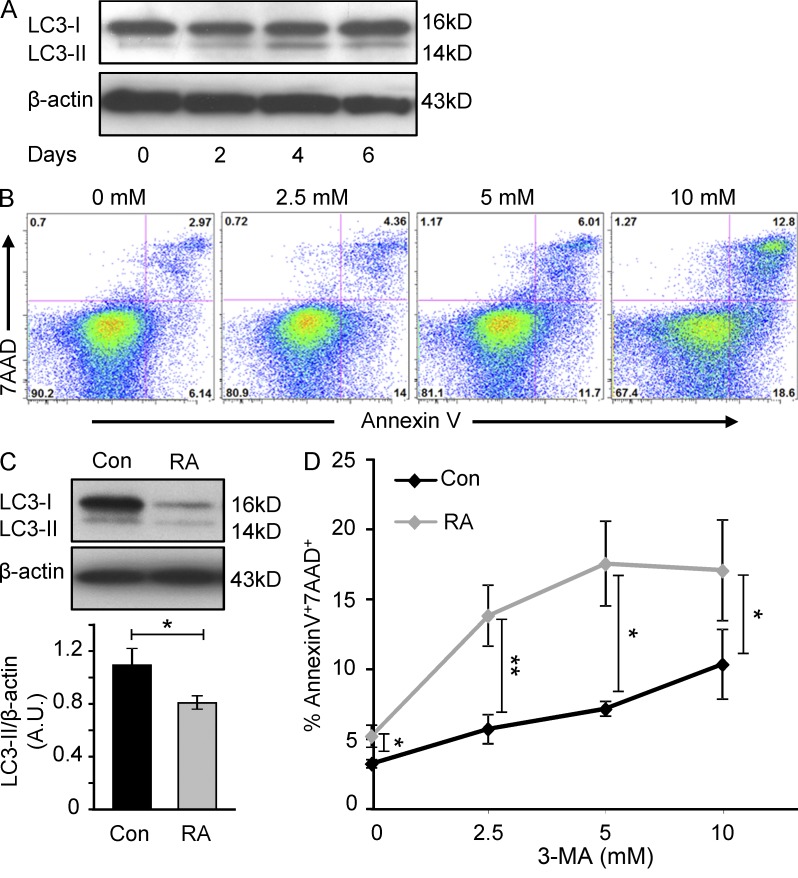
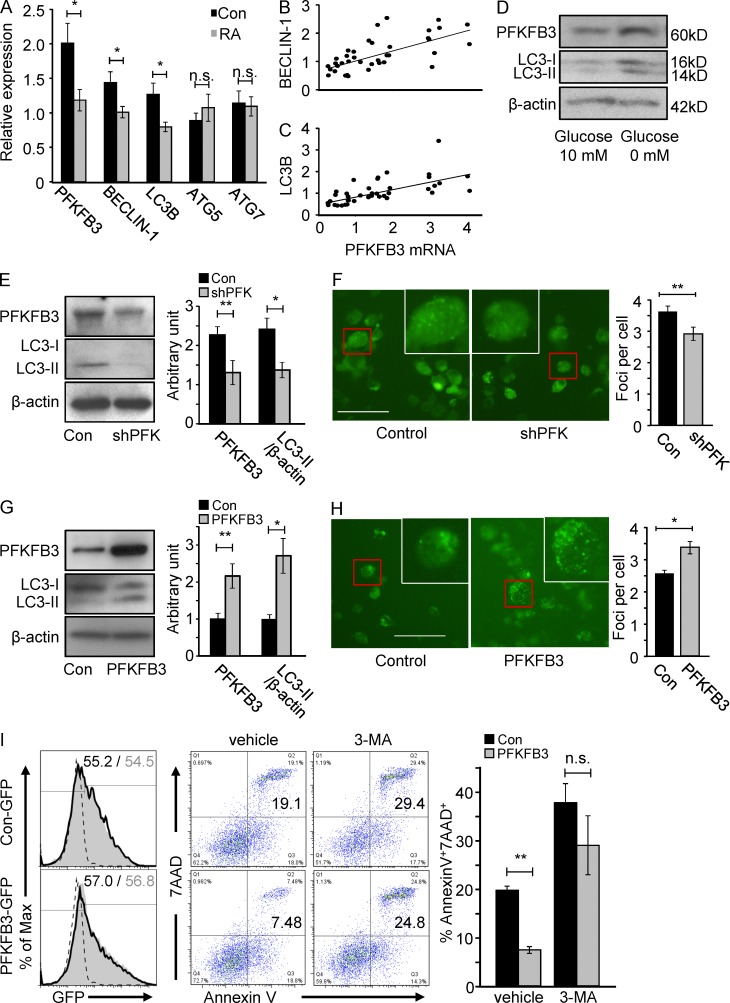
In the HLA class II-associated autoimmune syndrome rheumatoid arthritis (RA), CD4 T cells are critical drivers of pathogenic immunity. We have explored the metabolic activity of RA T cells and its impact on cellular function and fate. Naive CD4 T cells from RA patients failed to metabolize equal amounts of glucose as age-matched control cells, generated less intracellular ATP, and were apoptosis-susceptible. The defect was attributed to insufficient induction of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3), a regulatory and rate-limiting glycolytic enzyme known to cause the Warburg effect. Forced overexpression of PFKFB3 in RA T cells restored glycolytic flux and protected cells from excessive apoptosis. Hypoglycolytic RA T cells diverted glucose toward the pentose phosphate pathway, generated more NADPH, and consumed intracellular reactive oxygen species (ROS). PFKFB3 deficiency also constrained the ability of RA T cells to resort to autophagy as an alternative means to provide energy and biosynthetic precursor molecules. PFKFB3 silencing and overexpression identified a novel extraglycolytic role of the enzyme in autophagy regulation. In essence, T cells in RA patients, even those in a naive state, are metabolically reprogrammed with insufficient up-regulation of the glycolytic activator PFKFB3, rendering them energy-deprived, ROS- and autophagy-deficient, apoptosis-sensitive, and prone to undergo senescence.
Figures
Comment in
-
Rheumatoid arthritis: Metabolic reprogramming of T cells in RA.Nat Rev Rheumatol. 2013 Nov;9(11):635. doi: 10.1038/nrrheum.2013.158. Epub 2013 Oct 8. Nat Rev Rheumatol. 2013. PMID: 24100458 No abstract available.
References
-
- Atsumi T., Chesney J., Metz C., Leng L., Donnelly S., Makita Z., Mitchell R., Bucala R. 2002. High expression of inducible 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (iPFK-2; PFKFB3) in human cancers. Cancer Res. 62:5881–5887 - PubMed
-
- Bando H., Atsumi T., Nishio T., Niwa H., Mishima S., Shimizu C., Yoshioka N., Bucala R., Koike T. 2005. Phosphorylation of the 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatase/PFKFB3 family of glycolytic regulators in human cancer. Clin. Cancer Res. 11:5784–5792 10.1158/1078-0432.CCR-05-0149 - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- AI057266/AI/NIAID NIH HHS/United States
- AI044142/AI/NIAID NIH HHS/United States
- AR042547/AR/NIAMS NIH HHS/United States
- R01 AI044142/AI/NIAID NIH HHS/United States
- P01 HL058000/HL/NHLBI NIH HHS/United States
- R01 AG045779/AG/NIA NIH HHS/United States
- R01 AI108906/AI/NIAID NIH HHS/United States
- AI090019/AI/NIAID NIH HHS/United States
- R01 EY011916/EY/NEI NIH HHS/United States
- HL058000/HL/NHLBI NIH HHS/United States
- U19 AI057266/AI/NIAID NIH HHS/United States
- R01 AR042527/AR/NIAMS NIH HHS/United States
- EY011916/EY/NEI NIH HHS/United States
- U19 AI090019/AI/NIAID NIH HHS/United States
- R56 AI044142/AI/NIAID NIH HHS/United States
- R01 AI108891/AI/NIAID NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
