

Multiscaled exploration of coupled folding and binding of an intrinsically disordered molecular recognition element in measles virus nucleoprotein
- PMID: 24043820
- PMCID: PMC3791790
- DOI: 10.1073/pnas.1308381110
Multiscaled exploration of coupled folding and binding of an intrinsically disordered molecular recognition element in measles virus nucleoprotein
Abstract
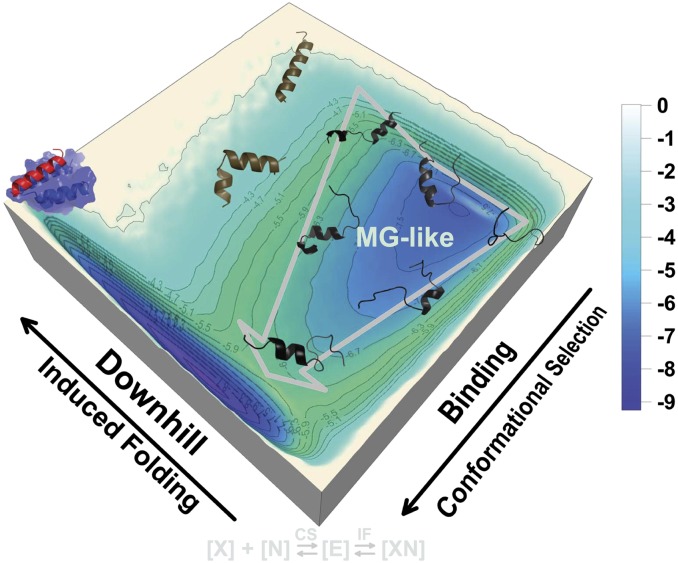
Numerous relatively short regions within intrinsically disordered proteins (IDPs) serve as molecular recognition elements (MoREs). They fold into ordered structures upon binding to their partner molecules. Currently, there is still a lack of in-depth understanding of how coupled binding and folding occurs in MoREs. Here, we quantified the unbound ensembles of the α-MoRE within the intrinsically disordered C-terminal domain of the measles virus nucleoprotein. We developed a multiscaled approach by combining a physics-based and an atomic hybrid model to decipher the mechanism by which the α-MoRE interacts with the X domain of the measles virus phosphoprotein. Our multiscaled approach led to remarkable qualitative and quantitative agreements between the theoretical predictions and experimental results (e.g., chemical shifts). We found that the free α-MoRE rapidly interconverts between multiple discrete partially helical conformations and the unfolded state, in accordance with the experimental observations. We quantified the underlying global folding-binding landscape. This leads to a synergistic mechanism in which the recognition event proceeds via (minor) conformational selection, followed by (major) induced folding. We also provided evidence that the α-MoRE is a compact molten globule-like IDP and behaves as a downhill folder in the induced folding process. We further provided a theoretical explanation for the inherent connections between "downhill folding," "molten globule," and "intrinsic disorder" in IDP-related systems. Particularly, we proposed that binding and unbinding of IDPs proceed in a stepwise way through a "kinetic divide-and-conquer" strategy that confers them high specificity without high affinity.
Keywords: flexible binding; flexible recognition; free-energy surface; hybrid structure-based model; multiscale simulation.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
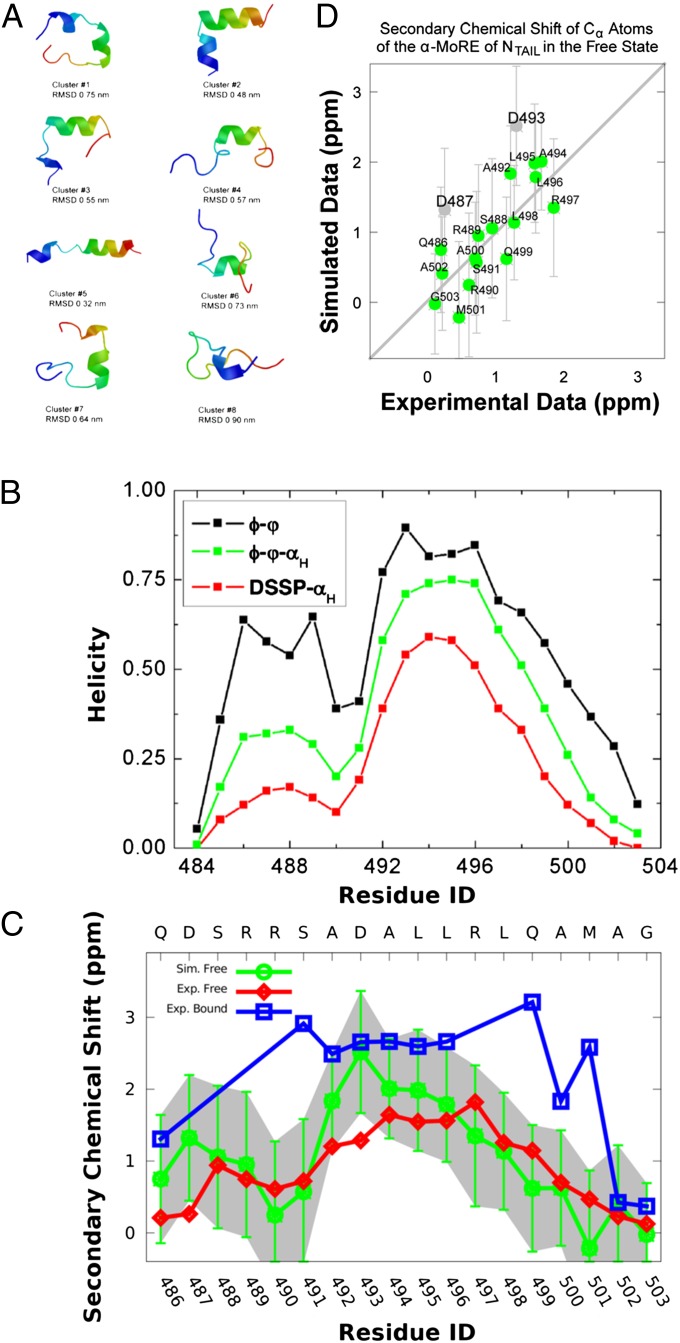
 chemical shifts for the α-MoRE. Green: experimental data of the free form. Red: simulated data of the free form from REMD simulation at 298 K. Blue: experimental data of the bound form at room temperature. Gray: estimated errors for the chemical shift prediction. (D) Correlation between simulated and experimental secondary chemical shifts with a correlation coefficient of 0.73. The gray points represent the residues, Asp487 and Asp493, which have relatively bad correlations.
chemical shifts for the α-MoRE. Green: experimental data of the free form. Red: simulated data of the free form from REMD simulation at 298 K. Blue: experimental data of the bound form at room temperature. Gray: estimated errors for the chemical shift prediction. (D) Correlation between simulated and experimental secondary chemical shifts with a correlation coefficient of 0.73. The gray points represent the residues, Asp487 and Asp493, which have relatively bad correlations.
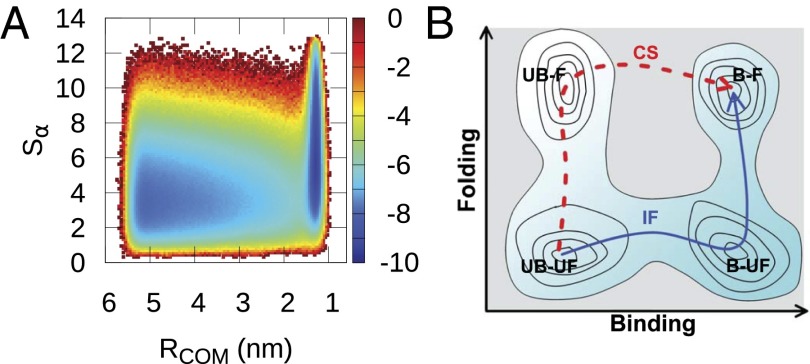
 (folding order parameter). Free-energy surfaces as a function of other folding order parameters and binding order parameters are shown in
(folding order parameter). Free-energy surfaces as a function of other folding order parameters and binding order parameters are shown in 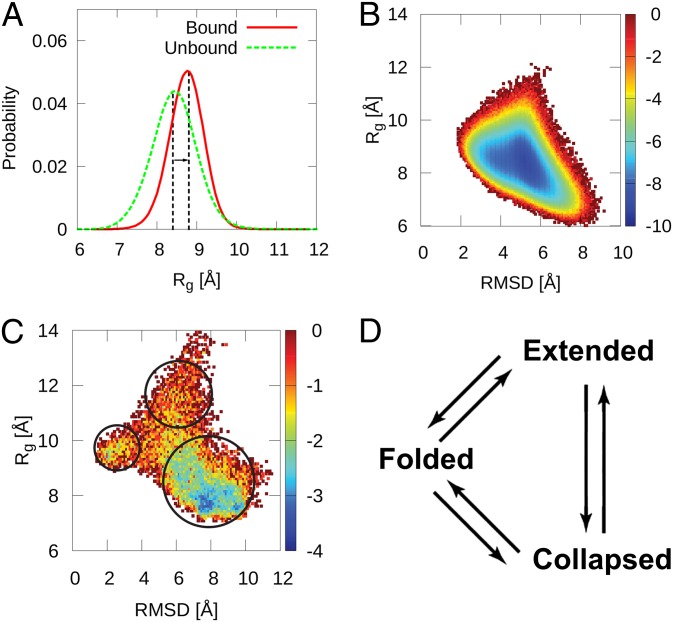
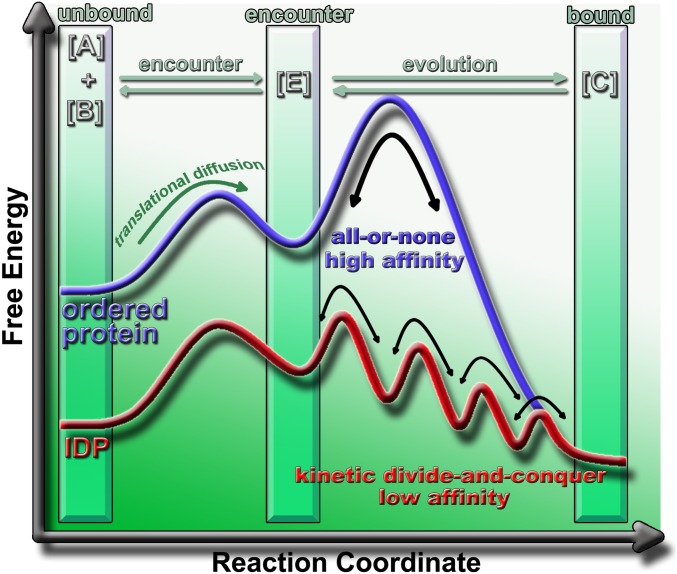
 . Here, [A] is an ordered protein or an IDP, [B] is a rigid partner of [A], [E] is the encounter complex, and [C] is the final bound complex. There are three states: an unbound state, an encounter state, and a bound state. In such three-state model, [A] has to encounter [B] to form [E] before evolving to form [C]. The encountering between [A] and [B] is under the control of translational and rotational diffusion. The major differences in the binding mechanism between ordered proteins and IDPs lie in the evolution step. IDPs form and break interactions with their partners in a gradual way (red) rather than according to an all-or-none discrete mechanism as observed for the docking of ordered proteins (blue). In this way, the association/dissociation of IDPs is determined by the low-energy barrier transitions and becomes relatively fast.
. Here, [A] is an ordered protein or an IDP, [B] is a rigid partner of [A], [E] is the encounter complex, and [C] is the final bound complex. There are three states: an unbound state, an encounter state, and a bound state. In such three-state model, [A] has to encounter [B] to form [E] before evolving to form [C]. The encountering between [A] and [B] is under the control of translational and rotational diffusion. The major differences in the binding mechanism between ordered proteins and IDPs lie in the evolution step. IDPs form and break interactions with their partners in a gradual way (red) rather than according to an all-or-none discrete mechanism as observed for the docking of ordered proteins (blue). In this way, the association/dissociation of IDPs is determined by the low-energy barrier transitions and becomes relatively fast.Comment in
-
Testing the validity of ensemble descriptions of intrinsically disordered proteins.Proc Natl Acad Sci U S A. 2014 Apr 22;111(16):E1557-8. doi: 10.1073/pnas.1323876111. Epub 2014 Mar 17. Proc Natl Acad Sci U S A. 2014. PMID: 24639541 Free PMC article. No abstract available.
-
Reply to Jensen and Blackledge: Dual quantifications of intrinsically disordered proteins by NMR ensembles and molecular dynamics simulations.Proc Natl Acad Sci U S A. 2014 Apr 22;111(16):E1559. doi: 10.1073/pnas.1400340111. Proc Natl Acad Sci U S A. 2014. PMID: 24877227 Free PMC article. No abstract available.
Similar articles
-
Simulation of coupled folding and binding of an intrinsically disordered protein in explicit solvent with metadynamics.J Mol Graph Model. 2016 Jul;68:114-127. doi: 10.1016/j.jmgm.2016.06.015. Epub 2016 Jul 1. J Mol Graph Model. 2016. PMID: 27423742
-
Mechanism of Coupled Folding-upon-Binding of an Intrinsically Disordered Protein.J Am Chem Soc. 2020 Jun 24;142(25):11092-11101. doi: 10.1021/jacs.0c03217. Epub 2020 Jun 16. J Am Chem Soc. 2020. PMID: 32323533
-
Demonstration of a folding after binding mechanism in the recognition between the measles virus NTAIL and X domains.ACS Chem Biol. 2015 Mar 20;10(3):795-802. doi: 10.1021/cb5008579. Epub 2014 Dec 22. ACS Chem Biol. 2015. PMID: 25511246
-
Binding induced folding: Lessons from the kinetics of interaction between NTAIL and XD.Arch Biochem Biophys. 2019 Aug 15;671:255-261. doi: 10.1016/j.abb.2019.07.011. Epub 2019 Jul 19. Arch Biochem Biophys. 2019. PMID: 31326517 Review.
-
The measles virus N(TAIL)-XD complex: an illustrative example of fuzziness.Adv Exp Med Biol. 2012;725:126-41. doi: 10.1007/978-1-4614-0659-4_8. Adv Exp Med Biol. 2012. PMID: 22399322 Review.
Cited by
-
Atomistic picture for the folding pathway of a hybrid-1 type human telomeric DNA G-quadruplex.PLoS Comput Biol. 2014 Apr 10;10(4):e1003562. doi: 10.1371/journal.pcbi.1003562. eCollection 2014 Apr. PLoS Comput Biol. 2014. PMID: 24722458 Free PMC article.
-
Designing disorder: Tales of the unexpected tails.Intrinsically Disord Proteins. 2013 Jan 1;1(1):e26790. doi: 10.4161/idp.26790. eCollection 2013 Jan-Dec. Intrinsically Disord Proteins. 2013. PMID: 28516025 Free PMC article. Review.
-
Modulation of Re-initiation of Measles Virus Transcription at Intergenic Regions by PXD to NTAIL Binding Strength.PLoS Pathog. 2016 Dec 9;12(12):e1006058. doi: 10.1371/journal.ppat.1006058. eCollection 2016 Dec. PLoS Pathog. 2016. PMID: 27936158 Free PMC article.
-
Investigating the Role of Large-Scale Domain Dynamics in Protein-Protein Interactions.Front Mol Biosci. 2016 Sep 13;3:54. doi: 10.3389/fmolb.2016.00054. eCollection 2016. Front Mol Biosci. 2016. PMID: 27679800 Free PMC article. Review.
-
Structural Disorder within Paramyxoviral Nucleoproteins and Phosphoproteins in Their Free and Bound Forms: From Predictions to Experimental Assessment.Int J Mol Sci. 2015 Jul 10;16(7):15688-726. doi: 10.3390/ijms160715688. Int J Mol Sci. 2015. PMID: 26184170 Free PMC article.
References
-
- Dunker AK, Obradovic Z, Romero P, Garner EC, Brown CJ. Intrinsic protein disorder in complete genomes. Genome Inform Ser Workshop Genome Inform. 2000;11:161–171. - PubMed
-
- Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6(3):197–208. - PubMed
-
- Chen J, Cheng J, Dunker AK. Intrinsically disordered proteins: Analysis, prediction, simulation, and biology. Pac Symp Biocomput. 2012;17:67–69.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
